








Servicios Personalizados
Articulo
 Articulo en PDF
Articulo en PDF Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
Links relacionados
 Citado por SciELO
Citado por SciELO
 Similares en SciELO
Similares en SciELO
Bookmark
Revista Cientifica Arte y Ciencia Medica
versión impresa ISSN 9999-8888
Rev. Arte y Ciencia Medica n.7 Sucre 2005
CASOS CLÍNICOS
Displasia fibrosa poliostótica a propósito de un caso
Poliostotic fibrous displasy presented through a case
AUTORES:Wendy Crespo Camargo
ASESOR: Dr. Fernando Carrasco Mérida
Resumen:
Presentamos un caso de Displasia Fibrosa Poliostótica, en una niña de 6 años de edad con un cuadro clínico de 3 años de evolución, que se inició con dolor a nivel de articulación de cadera izquierda, acortamiento de miembro inferior izquierdo y presencia de nódulos óseos indoloros a nivel de muslo izquierdo, la misma fué atendida en el servicio de Traumatología del Hospital Jaime Mendoza y diagnosticada mediante biopsia, TAC, y gamagrafía.
Palabras clave: Displasia Fibrosa Poliostótica.
Abstract:
We present a Poliostotic Fibrose Displasy case in a 6 year old girl which has been evoluing for the last three years. It started with pain at he left hip articulation level, left lower membe'r shortening and presence of painless osseous nodules in the left thigh level. She received medical treatment in the Trauma Department of Jaime Mendoza Hospital and diagnosticated through biopsy, TAC and gamagraphy..
Key words: Poliostotic Fibrose Displasy.
INTRODUCCIÓN
La Displasia Fibrosa, enfermedad descrita en 1938 por Lichtenstein, es definida por la OMS como: "un proceso benigno de naturaleza probablemente malformativa, caracterizado por la presencia de tejido fibroso conectivo con una disposición arremolinada característica y en las que se encuentra hueso maduro no laminar".
Puede ser monostótica o con menor frecuencia poliostótica según comprometa uno o varios huesos del esqueleto respectivamente. La presentación poliostótica se puede encontrar formando parte de síndromes definidos como el de McCune-Albright, en el que se acompaña de manchas cutáneas tipo café con leche y precocidad sexual femenina.
La etiología: es por la mutación en un gen en el cromosoma 20, lo que produce una alteración de una subunidad alfa de la proteína G, lo que se traduce en una alteración del medio extracelular cuando las células están en su desarrollo embriónico, y como producto de esto, los fibroblastos y osteoblastos tienen una función alterada. Además se ha visto que la mutación de esta proteína produce una alteración, donde la interleuquina -6 (que es inductora de la reabsorción ósea) está incrementada. Ocurre típicamente en la adolescencia, aunque una cuarta parte de las lesiones se dan en los adultos. Tiene un ligero predominio por las mujeres. Se presenta en niños de alrededor de 10 años de edad.
La distribución y extensión de las lesiones varía ampliamente, desde el compromiso de unos pocos huesos de una extremidad hasta la afectación de más del 50% de los huesos del esqueleto, en el 90% de los casos unilateral. Frecuentemente está implicada la pelvis, seguida de los huesos largos, cráneo, costillas, y extremidad proximal del fémur. Las lesiones tienen una apariencia agresiva, y la enfermedad progresa rápidamente, hasta que el esqueleto alcanza su madurez; después solo un 5% de las lesiones sigue creciendo. Los hallazgos incluyen dolor, fractura patológica, deformidad del miembro. En otras ocasiones la primera manifestación es una disfunción endocrina, (ales como acromegalia, hipertiroidismo, hiperpara-tiroidismo, síndrome de Cushing y pigmentación cutánea (manchas café con leche), constituyendo el síndrome de Albright-McCune. La complicación másfrecuente de la displasia fibrosa poliostótica es la fractura patológica.
DIAGNÓSTICO POR IMÁGENES:
Radiología.- La apariencia de las lesiones es inconstante y depende de la proporción de los componentes óseos y fibrosos de la lesión que ocupan grandes áreas en el interior del hueso. Si predomina el componente de tejido conectivo se verá una lesión diafisaria intramedular radiolúcida que se combina con adelgazamiento y abombamiento de la cortical.
Por el contrario si predomina el componente óseo el aspecto radiográfico será el de una lesión de vidrio esmerilado O nebuloso que puede asociarse con deformidad angular.
Esta forma se puede observar más frecuentemente en las lesiones de la base del cráneo y maxilares. En el nivel de lesión suele haber deformidad angular del hueso. La lesión activa puede progresar en el tamaño y deformidad.
Lesión tipo quístico: Radioopacidad con un margen reactivo, ninguna trabécula. y el espesor cortical normal. Lesiones tipo pagetoide: Patrón trabecular más denso que el hueso normal. Deformidad en cayado o bastón: La amplia afectación del fémur proximal produce una característica deformidad en varus que se parece a la curva de un bastón.
Gammagrafía: Es la forma más rápida para determinar la distribución de las lesiones esqueléticas, además de permitir descubrir lesiones en sitios insospechados. Revela intensa captación del radioisótopo que refleja la magnitud del tumor vista en la radiografía, especialmente cuando la lesión es activa. La disminución de la actividad en el scan óseo implica que la lesión se ha vuelto inactiva.
TAC y RNM: Puede ayudar a evaluar con exactitud la magnitud de la afectación ósea. La intensidad de señal en la RNM es moderadamente baja en TI, mientras en T2 es alta o media. Con gadolinio, la mayoría de las lesiones muestran un incremento central de contraste y algunos anillos periféricos. En general, la intensidad de señal depende de la cantidad de trabéculas óseas, colágeno, quistes y hemorragias.
HISTOLOGÍA:
Hay una colección irregular de trozos pequeños de tejido inmaduro trabecular rodeado por abundante proliferación fibroblástica de matriz de tejido fibroso y tejido trabecular inmaduro.
Algunas lesiones pueden contener cantidades grandes de cartílago benigno, mientras otros pueden tener componentes quísticos grandes. Histológicamente, los rasgos típicos incluyen hueso trabecular displásico truncado produciendo segmentos cortos del hueso, irregulares (llamados sopa del alfabeto chino). Las trabéculas displásicas están típicamente presentes dentro del estroma fibroso que reemplaza el hueso normal, y la actividad celular de estas lesiones es moderada. La apariencia se ha asemejado a sopa de letras, con trabéculas que se parecen a "C, O. U". Trabéculas: aparecen inmaduras, no están revestidas con osteoblastos, no contiene las líneas de cemento, no remodela según la tensión, el estroma fibroso es desorganizado y reemplaza a la médula normal.
TRATAMIENTO:
El tratamiento de la displasia fibrosa sigue siendo una tarea desafiante en niños y adultos. El paciente típico que tiene una lesión o poliostótica normalmente se beneficia de la fijación intramedular de ese segmento óseo. Se requiere una biopsia en la mayoría de las situaciones. El curetaje se asocia con una proporción muy alta de repetición local y por consiguiente, normalmente no se recomienda.
Así, se tratan típicamente mejor las lesiones de displasia fibrosa con biopsia seguida por algún tipo de injerto cortica] o fijación del implante para estabilizar el segmento del hueso largo. En el fémur, esto se logra con un injerto cortical de peroné o un clavo femoral encerrojado, dependiendo de la edad del paciente. El niño con una lesión grande tendrá una deformidad dolorosa progresiva, continuada, y pueden requerirse intervenciones múltiples. Esto es sobre todo cierto para los pacientes con enfermedad severa.
Cuando la lesión es muy desfigurante se puede tratar en forma radical haciendo la resección con un criterio oncológico y posteriormente rehabilitar al paciente.
Usando calcitonina, que podría ayudar a lo que es la remodelación ósea. También se puede usar Pamidronato (bifosfonato), que son medicamentos que impiden la reabsorción ósea, muy usada en pacientes con osteoporosis.
Actualmente la radioterapia está contraindicada porque hay una transformación sarcomatosa de la lesión. Estas lesiones una vez remodeladas tiene una alta recidiva, estos paciente cuando se les hace el remodelado deben seguir controlándose.
DIAGNÓSTICO DIFERENCIAL:
La Displasia Fibrosa Poliostótica entrará en el diagnóstico diferencial con la encondromatosis y la neurofibromatosis. En la encondromatosis a diferencia de la Displasia Fibrosa Poliostótica, las lesiones pueden extenderse en la parte articular del hueso y se extienden bandas radiolucentes del platillo de crecimiento a la metáfisis.
En la Neurofibromatosis la afectación esquelética normalmente se ve en deformidades en los hueos largos sin los cambios típicos intramedulares de la Displasia Fibrosa Poliostótica. Si la neurofibromatosis se asocia con fibromas no oscificantes múltiples (Síndrome de Campanacci), puede confundirse con la Displasia Fibrosa.
CASO CLÍNICO:
Paciente de sexo femenino de 6 años de edad, que acude a consulta externa de traumatología hace 2 años por presentar dolor a nivel de articulación de cadera izquierda.
EXAMEN FÍSICO:
Paciente, orientada en tiempo y espacio, facies tranquila, piel normotérmica, mucosas húmedas y rosadas.
A la inspección presenta acortamiento de miembro inferior izquierdo acompañado de trastornos en la marcha con evolución de aproximadamente 1 año; también se observa a nivel de muslo izquierdo, presencia de múltiples nódulos óseos indoloros a la palpación.
EXÁMENES COMPLEMENTARIOS:
RX: Indican presencia de zonas de apariencia quística en fémur derecho e izquierdo a nivel de trocánteres.
Gamagrafia: Tres fases con: Radiofármaco Te 99 m - MDP, previa administración del trazador por vía IV, estudio en fases vascular y tisular de pelvis y fémures, pool de cuerpo entero e imágenes tardías de rastro corporal total anterior y posterior, estáticas del tórax LI, OPI, pelvis PA y AP, piernas AP y PA fémur.
En la fase vascular y tisular se observa notable incremento de perfusión en articulación de cadera izquierda, metáfisis y diáfisis de fémur y tibia del mismo lado, sexto arco costal anterior y posterior izquierdos y decimosegundo arco posterior del mismo lado.
La fase ósea muestra distribución irregular del radiofármaco presentando intenso incremento difuso de captación en ala iliaca y rama isquiopubiana izquierda, articulación coxofemoral, masa trocantérea, metáfisis y diáfisis captación difusa en igual intensidad en sexto arco costal anterior y posterior, en decimo segundo arco costal posterior izquierdo.
La captación del radiofánnaco en ambos riñones está notablemente disminuida.
Biopsia:
Examen macroscópico: Izq.: Varios fragmentos de lx 0.8 cm; Der.: Varios fragmentos de 0.7 x 0.8 cm.
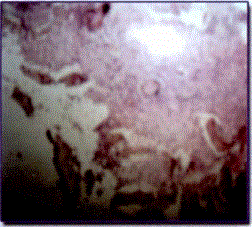
Examen microscópico: - Los cortes efectuados muestran tejido fibroconjuntivo con infiltrado linfocitario. Láminas óseas sin alteración.
- Los cortes efectuados muestran tejido fibroconjuntivo con abundantes núcleos pequeños e hipercromáticos. Láminas óseas con tendencia a la osificación.
DIAGNÓSTICO:
- Proceso inflamatorio crónico inespecífico
- Cambios celulares sugerentes de displasia
T.A.C. de Cadera:
Abdomen, pelvis, fémur sin contraste. Imágenes hipodensas con trabeculación hiperdensa a nivel de la pala iliaca izquierda con ruptura del cortex.
Deformación de la cabeza femoral y cuello del fémur izquierdo.
A nivel del hueco iliaco derecho: hipodensidad.
Compatible con osteocondromatosis Tumor de células gigantes?
EVOLUCIÓN DEL CASO.-
Se inician estudios de valoración pre operatoria para realizar punción y biopsia que no se concreta en esa oportunidad, porque la paciente es internada por presentar fractura patológica de tercio superior de fémur izquierdo, que es tratado con tracción e inmovilización quedando una angulación en dicha fractura.
El 8/XI /02 se interna en el servicio de pediatría para que se realice toma de biopsia de fémur derecho e izquierdo. Resultados de Anatomía Patológica: Proceso inflamatorio crónico inespecífico; cambios celulares sugerentes de displasia fibrosa ósea.
Ultrasonido abdomino pélvico:
Normal. Espacio infra y retroperitoneales normales.
TAC de abdomen y pelvis: Informa que se trata de osteocondromatosis - tumor de células gigantes.
Ante la duda, se realiza interconsulta con Oncología y una junta médica donde se determina que la paciente sea sometida a gamagrafía ósea.
En una nueva junta médica, con los resultados obtenidos de radiología, gamagrafía, anatomía patológica y una revisión bibliográfica actual, se llega al diagnóstico de Displasia Fibrosa Poliostótica.

COMENTARIO:
La displasia fibrosa ósea poliostótica, enfermedad poco frecuente que se caracteriza por una anomalía del desarrollo del mesénquima formador de hueso, afecta a personas jóvenes ( 1a- 2a década ), siendo el dolor y las fracturas patológicas la forma de presentación más frecuente.
Después de que cesa el crecimiento esquelético puede notarse un freno en el desarrollo de esta patología, incluso revertirse. En raras ocasiones puede haber transformación maligna así no haya antecedentes de irradiaciones previas.
Existen pocos casos de displasia fibrosa poliostótica, siendo un caso clínico poco frecuente, que en la mayoría de las veces simulan diversos cuadros clínicos, sin embargo minuciosos estudios radiológicos, histológicos, TAC, Gamagrafía y Resonancia Magnética nos brindaran el diagnóstico definitivo para posteriormente realizar el tratamiento específico correspondiente.
BIBLIOGRAFÍA:
1. Robbins S. Cotran R. Kumar V. Patología Estructural y Funcional. Ed. Mac Graw-Hill. 4 Ed (Vol II) 1990: Pag. 1399 - 1401 [ Links ]
2. Farreras P. Rozman C. Medicina Interna. Ed. Harcourts S.A. 14 Ed (Vol I) 2004: Pag. 1252 - 1253 [ Links ]
3. Behrman R. Kliegman R. Jen son H. Nelson Tratado de Pediatría. Ed. Elsevier. 17 Ed (Vol II) 2004: Pag. 1722 [ Links ]
4. Mahiques A. Displasia Fibrosa (05/07/2004) disponible en: www.arturomahiques.com/displasia_fibrosa.htm [ Links ]
5. Robaina L. Vega Ojeda A. Displasia fibrosa ósea como causa de fractura patológica en edad pediátrica. (18/02/04) disponible en: www.bvs.s1d.culrevistasiartivol l8_2_04/ortop07204 .htm. [ Links ]