








Serviços Personalizados
Artigo
 Artigo em PDF
Artigo em PDF Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
Links relacionados
 Citado por SciELO
Citado por SciELO
 Similares em SciELO
Similares em SciELO
Bookmark
Revista de Actualización Clínica Investiga
versão impressa ISSN 2304-3768
Rev. Act. Clin. Med v.45 La Paz jul. 2014
ARTICULO
Osteogenesis Imperfecta
Quelca Choque Heber Gonzalo1
Mg. Sc. Bustamante Cabrera Gladys2
1 Univ. Tercer Año Facultad de Odontología UMSA.
2 Médico Internista. Docente emérito UMSA. Mg.Sc. Psicopedagogía y Educación superior. Mg.Sc. Dirección de desarrollo Local. Mg.Sc. Planificación, gestión, evaluación de Proyectos. Mg. Sc. Bioética.
RESUMEN
La osteogénesis imperfecta es una enfermedad hereditaria caracterizada por formación defectuosa de los huesos a consecuencia de alteraciones en la síntesis de la producción de colágeno, proteína que se encuentra ausente o en muy escasa cantidad, principalmente en tejido óseo por lo que se produce una múltiple continuidad de fracturas en diferentes partes del cuerpo.
La osteogénesis imperfecta es un grupo de patologías, hereditarias que se dividen en seis tipos de cuadros, cada uno con una subclasificación que hacen de esta una patología compleja y de amplia distribución, donde el paciente recibe el gen que presenta disfunción de generar esta proteína responsable de dar solidez y firmeza a los tejidos duros del cuerpo.
La vida extrauterina del paciente cambia de manera significativa, incluyendo la de los padres, quienes juegan un papel preponderante en el desarrollo y adaptación del enfermo, enfrentando a la sociedad que tiende a mal interpretar con acusaciones de maltrato infantil a los que se encuentran en su entorno inmediato.
Los tratamientos actuales están dirigidos a prevenir y de alguna manera a controlar los síntomas de la enfermedad que a impedir que se desarrolle esta patología atacando directamente a la causa.
PALABRAS CLAVE
Osteogénesis Imperfecta. Colágeno. Densitometría. Huesos de cristal.
ABSTRACT
Osteogenesis imperfecta is a hereditary disease characterized by defective bone formation as a result of alterations in the synthesis of collagen, a protein that is absent or very small quantities, mainly in bone tissue so a multiple continuity occurs fractures in different body parts.
Osteogénesis imperfecta is a group of diseases, hereditary divided into six types of the disease, each with a subcalsification that make this a complex and widespread disease, where the patient receives the gene dysfunction presents generate this protein responsible give strength and firmness to the hard tissues of the body.
The extrauterine life for the patient changes significantly, including fpr both parents, who play a major role in the development and adaptation of the patient, facing the society that tends to misinterpret with allegations of child abuse to those found in their environment immediately.
Current treatments are aimed at preventing and somehow control the symptoms of the disease to prevent this disease develops directly attacking the cause.
KEYWORDS
Osteogenesis Imperfecta. Collagen. Densitometry. Brittle bone.
INTRODUCCION
La osteogénesis imperfecta es una enfermedad que ha sido descrita desde la antigüedad, encontrándose piezas momificadas, en el Museo de Historia Natural de Gran Bretaña, además de múltiples comunicaciones realizadas por diferentes autores desde 1788, recibiendo inicialmente el nombre de Síndrome de Volnik en honor al profesional que describió la patología y determinó el nombre con el que ahora se lo conoce.1
Este cuadro ambiguamente descrito durante muchos años, fue clasificado por Sillence en 1979, planteando evidencias clínicas y genéticas en cada caso, iniciándose de esta manera la búsqueda de los genes causantes de la enfermedad.1 En el 2006 se descubren formas recesivas de la enfermedad que hasta entonces se consideraba de herencia dominante, ampliándose la clasificación propuesta por Sillence.
ETIOLOGIA
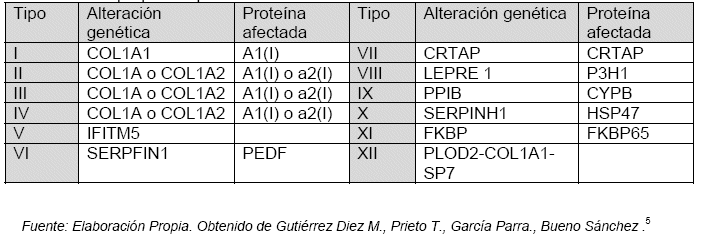
La enfermedad de los "huesos de cristal" u Osteogénesis imperfecta son un grupo de patologías genéticas hereditarias del colágeno que se caracteriza por presentar fragilidad ósea y alteraciones en otros órganos que tienen déficit de tejido conectivo, como los vasos sanguíneos, piel, uñas y dientes. Estas entidades requieren de un gen anormal, paterno o materno, para su presentación, es así que los genes identificados para su presentación son el COL1A1 y COL1A2 de los cromosomas 17 y 7 respectivamente, los cuales sufren una mutación que impide la codificación de las cadenas del colágeno, al momento de determinar las proteínas de la matriz de los tejidos duros y de otros componentes del cuerpo.2-3-4
Los genes y las proteínas afectadas en esta enfermedad son:
CLASIFICACION CUADRO CLINICO
Las características clínicas varían de un individuo a otro con Osteogénesis imperfecta en base a la clasificación realizada por Sillence.

A la clasificación de Sillence se añadieron otros tipos de osteogénesis imperfecta debido a que no todos los pacientes se ajustaban a las características clínicas de la enfermedad6, insertándose el:

También existe una clasificación que enmarca las características clínicas de dos tipos de la enfermedad, que consiste en una osteogénesis imperfecta congénita y otra tardía, la primera pertenece a los tipos II y III de la clasificación de Sillence caracterizándose por deformaciones y fracturas de los huesos luego del nacimiento, en tanto que la osteogénesis imperfecta tardía engloba a los tipos I y IV de la clasificación de Sillence.2
DIAGNOSTICO
El diagnóstico es básicamente clínico, en función a los hallazgos, perinatales y postnatales del individuo, debiéndose clasificar en los subtipos correspondientes en función a las características clínicas evidentes. De igual forma, la observación de las alteraciones genéticas confirmará el diagnostico de sospecha, así como la observación radiográfica de las lesiones. Radiográficamente se determinará la disminución en la densidad y matriz mineral de los huesos, múltiples fracturas, presencia de matrices exuberantes, alteraciones metafisiarias, etc. La biopsia de tejido epitelial y ósea coadyuvará de igual forma en el diagnóstico, acompañado de cultivos de fibroblastos.2
TRATAMIENTO
No existe tratamiento para la enfermedad, por lo que la prevención de las fracturas es el manejo más apropiado, apoyado con terapia física y soportes óseos como férulas ortodoncia, o silla de ruedas. El manejo quirúrgico solamente está indicado en la reparación de las lesiones o la corrección de las mismas, apoyado con el uso de medicamentos que fortalezcan la matriz ósea, como la hormona de crecimiento o bifosfonatos, de los cuales el pamidronato es el de mayor uso.3,9
El futuro del tratamiento en esta enfermedad, está enfocado a la corrección del gen alterado, a través de la terapia de supresión antisentido, que tiene como función disminuir la expresión del RNA mensajero, o el trasplante alogénico de médula ósea, que tienen como función la proliferación de un hueso normal, que si bien son terapias en desarrollo, pretenden ser usadas en un corto plazo.9
BIBLIOGRAFIA
1. Sanjurjo M .Características Genéticas de la Osteogénesis Imperfecta. 2010: 3-5. URL disponible en: http://www.osteogenesis.info/almacen/sanjurjo.pdf. Accedido en fecha 11 de junio de 2014 [ Links ]
2. Gracia, R; I González. Tratamiento de la Osteogénesis Imperfecta. Symposium: Avances y controversias en endocrinología pediátrica. Madrid-España. 2002:72-74. URL disponible en: http://www.seep.es/privado/documentos/congresos/C2002/11.pdf. Accedido en fecha 23 de mayo de 2014. [ Links ]
3. Salud: Osteogénesis Imperfecta. Technosite/Fundación ONCE. Madrid-España. 2009. URL disponible en: http://salud.discapnet.es/Castellano/ Salud/Enfermedades/EnfermedadesDiscapacitantes/O/Osteogenesis%20 Imperfecta/Paginas/cover%20osteog enesis.aspx. Accedido en fecha 26 de mayo de 2014. [ Links ]
4. Osteogénesis Imperfecta. Asociación Nacional de Huesos de Cristal. Madrid- España. 2012. URL disponible en: http://www.ahuce.org/Osteogenesisimperfecta/Tratamientos_en_Osteoge nesis_imperfecta.aspx. Visitado el 27 de mayo de 2014. [ Links ]
5. Gutiérrez M, Molina M, Prieto L, Parra J, Bueno A. Osteogénesis Imperfecta: Nuevas Perspectivas. Endocrinología Pediátrica. España. 2013: 76-78. URL disponible en: http://www.ahuce.org/Portals/0/Publicaciones/Aspectos_Generales/osteogenesis_imperfecta_Nuevas_Perspect ivas.pdf. Accedido en fecha 8 de junio de 2014. [ Links ]
6. Valenzuela G, Zárate H. Osteogénesis imperfecta: caso clínico y actualización. Revistas bolivianas. Bolivia. 2007. 1-4. URL disponible en: http://www.revistasbolivianas.org.bo/ pdf/chc/v52n1/v52n1a11 .pdf. Visitado el 31 de mayo de 2014. [ Links ]
7. García A. Actualización en Osteogénesis Imperfecta. URL disponible en: http://www.sld.cu/galerias/pdf/sitios/w illiamsoler/actualizacion_en_osteoge nesis_imperfecta.pdf. Visitado el 26 de mayo de 2014. [ Links ]
8. Asociación Madrileña de Osteogénesis Imperfecta. España. 2011. URL disponible en: http://www.sld.cu/galerias/pdf/sitios/w illiamsoler/actualizacion_en_osteoge nesis_imperfecta.pdf. Visitado el 31 de mayo de 2014. Caleya A, Altamirano L. La Dentinogénesis Imperfecta como alerta de Osteogénesis Imperfecta. Gaceta Dental. Madrid-España. 2011. URL disponible en: http://www.gacetadental.com/2011/09/la-dentinognesis-imperfecta-como-alerta-de-osteognesis-imperfecta-25497/. Accedido el 29 de mayo de 2014. [ Links ]