








Serviços Personalizados
Artigo
 Artigo em PDF
Artigo em PDF Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
Links relacionados
 Citado por SciELO
Citado por SciELO
 Similares em SciELO
Similares em SciELO
Bookmark
Revista de Actualización Clínica Investiga
versão impressa ISSN 2304-3768
Rev. Act. Clin. Med v.25 La Paz nov. 2012
ARTICULO
GRANULOMA CENTRAL DE CÉLULAS GIGANTES DEL SENO MAXILAR.
A PROPOSITO DE UN CASO
Dr. Aillón Huáscar 1
Dra. Gutiérrez Ana 2
RESUMEN
El granuloma central de células gigantes es una lesión osteolítica localizada, de naturaleza variablemente agresiva que afecta a los maxilares, se presenta con relativa frecuencia en pacientes pediátricos y jóvenes, en el presente artículo se presenta un caso clínico de granuloma central de células gigantes (GCCG) con seis meses de evolución, localizado en región geniana derecha, iniciándose en el seno maxilar derecho (presentando inclusión del canino sup. derecho), en una joven de veinticuatro años de edad, cuya lesión llegó a producir asimetría facial, mismo que se trató quirúrgicamente, eliminándolo por medio de excéresis.
PALABRAS CLAVE
Granuloma central de células gigantes (GCCG), lesión osteolítica.
INTRODUCCIÓN
Pseudotumor frecuente de la región dentada de los maxilares, producido posiblemente por reparación de procesos hemorrágicos, formado por tejido fibroso rico en células, donde se encuentra acúmulos focales de células gigantes multinucleadas.1
Las lesiones mesenquimatosas benignas representan el grupo más frecuente de tumores no odontogénicos en pacientes jóvenes. Se incluyen en este grupo: 1) lesiones de células gigantes, 2) lesiones fibroóseas y 3) mixomas. La tumefacción constituye el síntoma inicial característico en las tres clases de tumores, el dolor, los dientes desplazados o móviles y la resorción radicular acontecen con mayor frecuencia en personas con lesiones de células gigantes. Este tipo de padecimientos incluye granuloma central de células gigantes GCCG, tumor pardo de hiperparatiroidismo y tumor de células gigantes.4
Los tres son indistinguibles mediante técnicas histológicas estándar. Las células gigantes encontradas en estas formaciones se presentan en una variedad de tumores benignos y malignos de los maxilares: como quiste óseo aneurismático, displasia fibrosa, querubismo, fibroma osificante o sarcoma osteógeno.
La incidencia máxima del GCCG ocurre de 10 a 25 años de edad, es una lesión de relativa frecuencia que ocurre predominantemente en niños, adolescentes y jóvenes, 60 de 75% de los casos estudiados son diagnosticados en pacientes menores de 30 años.
La proporción entre mujeres y hombres es de 3:2, predominando en sexo femenino, y la lesión se presenta más a menudo en el maxilar inferior. El GCCG rara vez se encuentra en sentido posterior con respecto al primer molar permanente, por lo general, no provoca síntomas y se descubre por accidente en radiografías de rutina. Los síntomas pueden abarcar tumefacción local casi siempre sin dolor o parestesia, y el hallazgo inicial es una asimetría facial leve, engrosamiento del hueso afectado, trastornos de la respiración y o una repentina malposición de los dientes, o perdida de la oclusión, como también un sorpresivo aflojamiento de dientes que suelen permanecer vitales.
Generalmente se mantiene circunscrito al hueso afectado, y se manifiesta abombado, quedando siempre respetada su cortical.1
En el maxilar superior puede ocupar todo el seno maxilar e inclusive levantar el piso de la órbita (exoftalmo) e invadir fosas nasales (dificultad respiratoria). Solo ocasionalmente y también en caso de una anodoncia, se ha observado su protrusión dentro de la boca, apareciendo como un tumor redondo, violáceo, con tendencia sangrante, con las mismas características morfológicas de un épulis gigantocelular.1
Desde el punto de vista radiográfico, se observa una imagen radiolucida que por lo general se presenta relativamente grande con una línea de demarcación poco definida en relación al hueso normal, pudiendo tener incluido uno o varios dientes cuyos ápices pueden estar reabsorbidos. La expansión de las corticales vestibular y palatina o lingual suele observarse mediante la radiografía oclusal, las cuales presentan a menudo una ausencia completa del hueso cortical, comúnmente se observa desplazamiento de los dientes asociados.2
La existencia de un verdadero tumor de células gigantes de los maxilares es motivo de polémica; sin embargo, este tipo de células en los maxilares, es muy agresiva in situ con un índice de recurrencia elevado pero poca tendencia a desarrollar metástasis. Los tumores de células gigantes de los huesos largos se presentan en grupos de mayor edad, mientras que las lesiones agresivas de células gigantes en los maxilares se presentan en niños y jóvenes. Los tumores de los huesos largos son más destructivos, sufren recidiva más a menudo; entre 15 a 30% se tornan malignos con tendencia a sufrir metástasis en los pulmones; en consecuencia, es probable que ambos tumores sean distintos.5
Se han clasificado las lesiones de GCCG en dos categorías: 1) agresivas y 2) no agresivas, mediante técnicas radiográficas y citométricas con la finalidad de aportar más datos acerca de esta patología. Las lesiones agresivas de células gigantes tienden a ocurrir en pacientes pediátricos, con una edad promedio de seis años, provocan dolor, tienen crecimiento rápido y de modo característico muestran resorción radicular y perforación de la corteza en las radiografías. Las lesiones no agresivas de células gigantes se presentan en una edad promedio de nueve años, radiográficamente se aprecian como zonas radiolúcidas que no causan dolor, no se observa resorción radicular o perforaciones de las corticales óseas. Las lesiones agresivas poseen un índice de recurrencia alto a menos que se traten mediante extirpación en bloque.4
La terapéutica de las lesiones de células gigantes depende de la combinación de las características clínicas e histológicas, y más importante, el comportamiento histológico de la lesión. El curetaje es el tratamiento primario conveniente para el GCCG no agresivo. Posiblemente se requiera la extirpación de lesiones recurrentes, luego de descartar hiperparatiroidismo. La resección en bloque es la mejor terapéutica en lesiones agresivas de células gigantes con crecimiento rápido, dolor, expansión ósea, perforación, resorción radicular. Existe un método alternativo de tratamiento no quirúrgico para el GCCG, el cual consiste en la administración intralesional de corticoesteroides, según un estudio por Jacoway y colaboradores, en donde se ha podido observar regeneración ósea en las áreas osteolíticas y disminución del edema facial, el medicamento utilizado es triamcinolona 10 mg mezclado con xylocaína al 0.5%, inyectado dentro de la lesión 2 ml. de la solución por cada dos cm. de su tamaño, aplicada semanalmente por seis semanas aproximadamente.4
La extirpación quirúrgica, asociada o no a otra terapia, adyuvante es el tratamiento de elección en la mayoría de los casos. Las vías de abordaje empleadas dependerán de la localización, tipo, extensión y comportamiento biológico del granuloma, eligiendo aquel que nos permita una adecuada exposición para realizar una resección segura y satisfactoria.3
Finalmente, subrayamos la importancia de la primera visita, del diagnóstico radiográfico e histológico para prevenir el posible daño de los dientes y del hueso adyacente.
CASO CLÍNICO
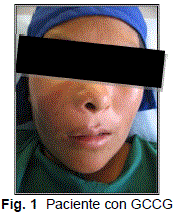
Se realizó una valoración clínica, un análisis histológico y anatomopatológico con la finalidad de determinar diagnóstico.(Fig.1).
VALORACIÓN CLÍNICA
Anamnesis
Mujer boliviana de 24 años de edad, que responde a las iniciales B. CH. A., concubinada, ama de casa, sin antecedentes personales de interés o alergias a medicamentos conocidas.
La lesión presenta una evolución de seis meses aproximadamente, la cual estaba ubicada a nivel del surco nasogeniano derecho, de 1 cm aproximadamente, doloroso; con el transcurso de los días fue aumentando de volumen paulatinamente hasta sus dos meses de evolución; posteriormente aumento de volumen en forma acelerada en región geniana, nasal y labial derechas a expensas del maxilar superior.
Examen físico
Aumento de volumen en región geniana, nasal y labial superior derechas, de dimensión aproximada de 7.3 x 5.9 cm, duro y doloroso a la palpación, provocando asimetría facial importante (Fig.2), intraoralmente se palpa masa dura borrando parcialmente el fondo de surco vestibular derecho.

Examen Radiográfico
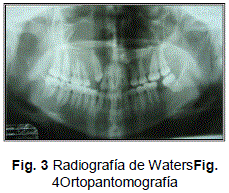
Para tener un mejor estudio del caso clínico se tomaron en cuenta dos radiografías:
Radiografía de Waters (Fig. 3 ) y la Ortopantografía (Fig. 4).El cuadro radiográfico muestra en el maxilar superior, en la región del antro del seno maxilar, un gran aumento de grosor del hueso con una estructura opalina amplia, con una imagen radiolúcida, la cual se encuentra poco delimitada, y se puede observar en su interior un canino incluido.

Tratamiento
En este caso clínico se realizó el abordaje mediante la incisión "Rinotomía con ampliación subciliar".
Procedimientos quirúrgicos:
Paciente en decúbito dorsal pasivo bajo efectos de anestesia general.
Se realizó la asepsia y antisepsia de región operatoria.
Se colocó los campos según técnica. Incisión de Weber - Ferguson Dieffenbach (Rinotomía lateral ampliada) (Fig. 5), esta incisión se inicia en el labio superior, en la línea media hasta el suelo de la fosa nasal de la columela. En este punto la incisión se hace lateral y cefálica para entrar en el suelo de la cavidad nasal. Después de girar 45° saliendo del suelo de la fosa nasal se sigue el ala nasal y el borde lateral de la nariz y se asciende lateralmente al dorso nasal hasta el nivel del canto interno3

Posteriormente se procedió a la diéresis aguda con bisturí, por planos, se identifica granuloma, se observóla presencia del canino superior derecho incluido, con ausencia de pared anterior de seno maxilar, el GCCG abarcaba hasta piso de órbita, fosa pterigomaxilar, fosa nasal derecha y la totalidad de seno maxilar, se realizó hemostasia compresiva y electrocoagulación lavando intermitentemente el lecho operatorio.
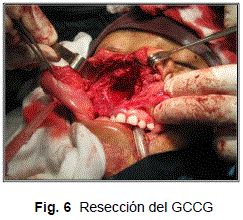
Se resecó el GCCG en su totalidad (Fig. 6), revisando minuciosamente lecho operatorio. Inmediatamente después se aplicó un tapón hemostático con gasas impregnadas con yodoformo.

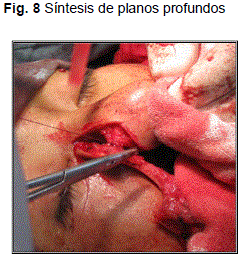
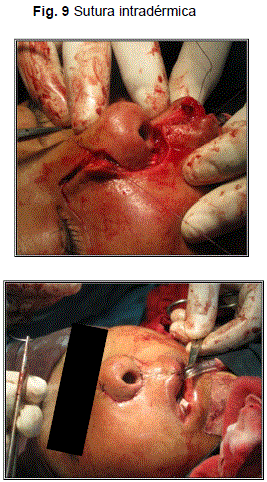
Posteriormente se procedió a la síntesis con hilo reabsorbible 3/0, puntos sueltos en planos profundos (Fig. 7-8); hilo nylon 5/0, puntos sueltos; y puntos continuos intradérmicos en piel (Fig. 8).

Histopatología
El estudio anatomopatológico confirmó el diagnóstico del granuloma central de células gigantes, descrito como tejido fribroconjuntivo con células gigantes multinucleadas tipo cuerpo extraño, numerosos capilares sobre todo en la periferie de la lesión, un constituyente más de la lesión es la presencia de espículas óseas, con hueso residual laminar que puede ser atrapado dentro de la lesión o puede ser visto como una capa sobre su periferia.
En la forma más agresiva del GCCG, histológicamente las células se observan más grandes y tienen más núcleos que la forma no agresiva, las divisiones mitóticas son más numerosas y frecuentes sin ser anormales.
Las características histológicas pueden ser idénticas para el tumor de células gigantes del esqueleto estomatognático o para el GCCG, así que sin importar la localización de la lesión el diagnóstico histológico puede ser también tumor de células gigantes de hueso.
El diagnóstico de la histopatología reportó: Granuloma central de células gigantes del seno maxilar.
Controles postoperatorios
En el postoperatorio inmediato la paciente refirió haber pasado la noche tranquila, con leve dolor a nivel de la herida operatoria, que cedió favorablemente.
Al examen físico presenta la hemicara derecha con ligero aumento de volumen, doloroso a la palpación, con presencia de dren laminar en cavidad oral, piel normotérmica, normohidratada. Se indica alta hospitalaria.
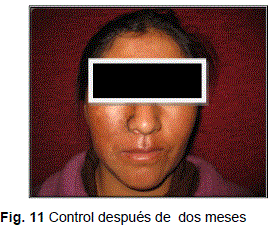
La paciente tuvo un postoperatorio mediato y tardío con evolución favorable (Fig. 11).A los dos meses, la paciente presenta evolución favorable; al examen físico, simetría facial, piel normotérmica y normohidratada, ausencia de dolor a la palpación
CONCLUSIÓN
El granuloma central de células gigantes es una afección que aún no se sabe su etiología, es poco frecuente (más aún en el maxilar superior) por lo cual existe escasa evidencia y poca experiencia en esta patología.
El estado funcional, el compromiso de estructuras vitales y o estéticas del individuo, que padece de esta patología, determina el tipo de tratamiento, si éste será farmacológico o quirúrgico por lo cual se determinará el abordaje y extensión de la resección.
Por los antecedentes, cuadro clínico, evolución y el diagnóstico histológico se debe realizar el diagnóstico diferencial con lesiones de células gigantes, lesiones fibroóseas y mixomas. La tumefacción constituye el síntoma inicial característico en las tres clases de tumores, el dolor, los dientes desplazados o móviles y la resorción radicular acontecen con mayor frecuencia en personas con lesiones de células gigantes.
NOTAS
1 Odontólogo. Especialista en Cirugía MÁxilo Facial UMSA-UMRPSFX. Cirujano Maxilofacial. Especialista en labio Leporino La Paz-Bolivia
2 Odontóloga. UMRPSFX
BIBLIOGRAFÍA
1. Sandner Montilla O. Tratado de Cirugía Oral y Maxilofacial. Amolca. Año 2007; 615 y 616. [ Links ]
2. Sapp J. P. Eversale L. Wysoki G. Patología Oral y Maxilofacial Contemporánea. Mosby 2da Edición 119 y 120. [ Links ]
3. Carlos Navarro Vila, F. García Marin, S. OchandiaroCaicoya. Tratado de Cirugía Oral y Maxilofacial. Tomo III. Aran. 2004:1324 y 1325. [ Links ]
4. Corbeto T. Manejo de cirugías URL disponible en: www.odontochile.cl/archivos/tercero/.../gcentraldecelulasgigantes.ppt [ Links ]
5. Contreras Vidaurre E. Alternativa de tratamiento para el granuloma central de células gigantes URL disponible en: www.tesis.ufm.edu.gt/pdf/2455.pdf [ Links ]^rND^sFuentes De la Barra^nPaola Yocelyn^rND^sFuentes De la Barra^nPaola Yocelyn^rND^sFuentes De la Barra^nPaola Yocelyn
ARTICULO
SALICILATOS
Fuentes De la Barra Paola Yocelyn1
RESUMEN
Realizando una serie de investigaciones se descubrió que la corteza del sauce blanco tiene propiedades analgésicas y antipiréticas, dando lugar al descubrimiento de los AINES, clasificándose dentro de este grupo a los salicilatos.
Los salicilatos son los fármacos más antiguos que se conocen, ya nuestros antepasados utilizaban el sauce que era muy útil para tratar la fiebre, y la corteza del álamo que contenía un glúcido del alcohol salicílico que servía en ese entonces para tratar el dolor, fiebre, infecciones y gota. De esta manera los salicilatos son utilizados principalmente por la actividad analgésica que poseen; presentan también propiedades antiinflamatorias y antipiréticas actuando de esta forma en los casos de dolor, fiebre e inflamación de una manera significativa, ya que estos datos clínicos son la expresión de los mecanismos de defensa del organismo, los cuales provocan en el ser humano una experiencia sensorial y emocional desagradable, por lo que el ser humano a lo largo del tiempo ha tratado de combatirlo.
Los beneficios que presentan estos fármacos, son importantes, al igual de los efectos adversos por su uso inadecuado o indiscriminado.
PALABRAS CLAVES
Ácido acetil salicílico. Salicilatos. Diflunisal. Dolor
INTRODUCCION
A principios del año 1800 se descubrió el componente activo de los salicilatos, la salicilina, un glúcido del alcohol salicílico que fue sintetizado por primera vez el año 1827; el año 1830 se descubrió el ácido acetil salicílico, el cual es recién sintetizado por Gerhard el año 1853, posteriormente Dreser en 1989 decidió introducirlo como fármaco con el nombre comercial de aspirina, finalmente se fueron sintetizando otros compuestos de los salicilatos como el salicilato de metilo que se descubrió a través de las plantas de hoja de perene, el cual es elingrediente activo de muchos ungüentos cutáneos que se utilizan con propósitos analgésicos; el salicilato de sodio, el salicilato de colina y magnesio y el diflunisal.1,2,3,4.
De ésta manera los salicilatos constituyeron y constituyen un grupo de fármacos importantes utilizados sobre todo por las propiedades analgésicas que presentan, puesto que el ser humano lo empleaba y lo emplea a menudo debido a esta propiedad en particular.4
MECANISMO DE ACCION DE LOS SALICILATOS
El principal mecanismo de acción de los salicilatos es la inhibición de la actividad de la ciclooxigenasa, siendo ésta la enzima responsable de la conversión de ácidos araquidónicos en endoperóxidos, mismos que posteriormente se transforman en prostaglandinas y tromboxanos que son los eicosanoides responsables del dolor, fiebre e inflamación.5-7
FARMACOCINETICA
Administrados por vía oral son absorbidos rápidamente en el estómago e intestino delgado, llegando a su acción máxima a la hora de su administración y una vez en el plasma se combina con la albúmina para metabolizarse en el hígado a través de las mitocondrias hepáticas; su excreción se realiza vía renal mediante los mecanismos de filtración glomerular, reabsorción tubular y secreción tubular.5,8,9.
Administrados por vía rectal la absorción de los salicilatos es más lenta e irregular debido a que el fármaco se disuelve lentamente por la acción de la temperatura del organismo, en caso de que los comprimidos tengan cubierta entérica la absorción también se hace lenta debido a que el fármaco se disuelve lentamente en el estómago sufriendo posteriormente su metabolización en el hígado.3,5.
1. Acción analgésica: Los salicilatos tienen una acción analgésica del 30 %, en razón de que actúan solo a nivel de la reducción del umbral del dolor, mientras que la acción analgésica brindada por los analgésicos hipnóticos logran un 100% de analgesia por actuar sobre los tres componentes del dolor, que son, el umbral de la percepción dolorosa, la reacción psicológica y la depresión del sistema nervioso central como sucede en el caso de la morfina.1,4,7.
Los salicilatos presentan una acción analgésica a:
1. nivel periférico, por su acción directa sobre la inflamación, que al bloquearla limitan también el dolorporque inhiben los estímulos del mismo a nivel cerebral subcortical.5
2. nivel central, por la depresión del tálamo óptico centro principal de la percepción dolorosa, siendo la acción analgésica eficaz en la cefalea, artralgia, mialgia y odontalgia.8,9
Además presentan leve acción sedante del sistema nervioso central, con depresión de la corteza subcortical y reducción de COX 2.3
2. Acción antipirética:Los salicilatos reducen la temperatura corporal en personas con estados febriles, produciendo vasodilatación cutánea, disipando el calor por sudoración y evaporación; siendo entre los salicilatos el Diflunisal el que posee la mayor actividad antipirética, al cual le sigue, la aspirina y el salicilato de sodio , éstos últimos utilizados sobre todo en fiebre reumática. La acción antitérmica se debe a que producen un descenso sustancial de la temperatura actuando sobre el centro termorregulador del hipotálamo deprimiéndolo, es por eso, que los salicilatos poseen el mismo tipo de acción desde un punto de vista cualitativo y no así desde un punto de vista cuantitativo debido a que un fármaco puede presentar o no presentar alguna propiedad de mayor o menor actividad que otro fármaco.4,5,7
3. Acción antiinflamatoria: La acción antiinflamatoria que poseen los salicilatos se debe principalmente a la inhibición de prostaglandinas, cininas y peroxidasas, que al disminuir la formación del ácido hidroxieicosatetraenoico se bloquea la inducción de células inflamatorias, que llevan a tres fases importantes que son:
a. Fase aguda la cual se manifiesta por un aumento de la permeabilidad capilar.
b. Fase subaguda en la cual intervienen los linfocitos y la
c. Fase crónica la cual se caracteriza por ser degenerativa y fibrocítica. Los salicilatos actúan sobre estos agentes constituyéndose una acción importante de estos fármacos ya que al actuar sobre la inflamación también reducen el umbral del dolor. Esta acción es muy importante en el tratamiento de enfermedades las cuales impliquen las patologías como ser: artritis reumatoide y fiebre reumática por que reducen el dolor y la inflamación articular retardando el daño que podrían provocar los tejidos inflamados disminuyendo la permeabilidad capilar de los mismos3,5,6,9
4. Acción sobre el sistema cardiovascular: Los salicilatos con compuestos de sodio a dosis elevadas pueden precipitar un cuadro de insuficiencia cardiaca.2,4.
5. Acción en la respiración y equilibrio acido base: estimulan el centro respiratorio, siendo capaces de antagonizar la acción depresora respiratoria de la morfina.2-5.
6. Acción sobre la sangre: pueden inhibir la agregación plaquetaria, aumentando el tiempo de sangrado debido a su capacidad de acetilación hacia las membranas plaquetarias prolongando así el sangrado.2,4,7
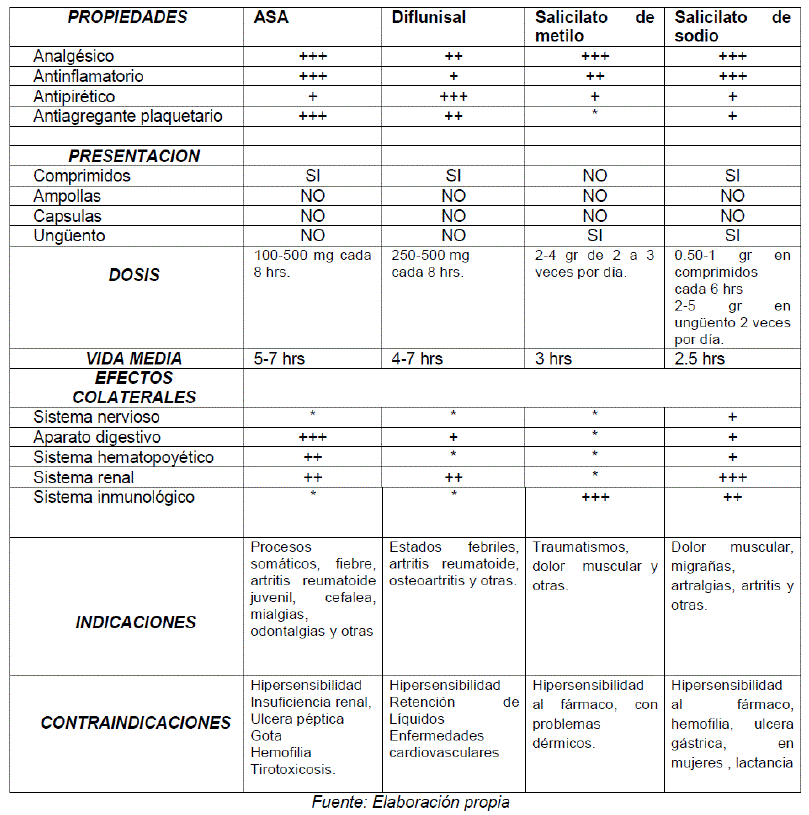
CLASIFICACION
Los fármacos que se encuentran dentro la clasificación de los salicilatos es:
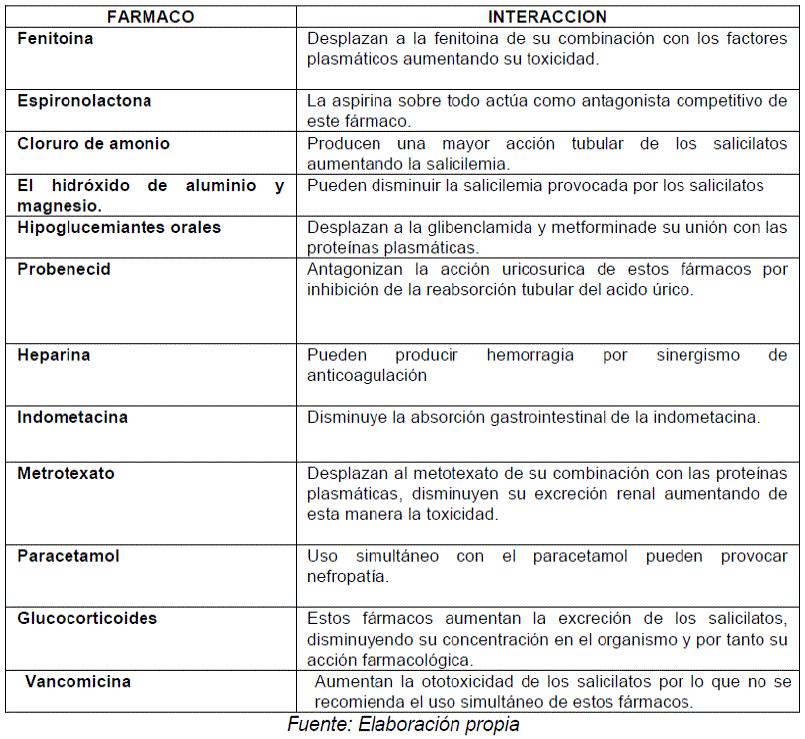
INTERACCIONES DE LOS SALICILATOS CON OTROS FARMACOS:
Las interacciones son:
TOXICIDAD DE LOS SALICILATOS
La intoxicación por salicilatos tiene la siguiente clasificación: toxicidad leve o crónica también conocida como salicilismo debido al consumo de cantidades excesivas, presentándose, así cefalea, mareos, visión borrosa, confusión mental, anemia ferropénica; toxicidad grave o aguda presentándose convulsiones generalizadas, coma, erupciones cutáneas, alteraciones del equilibrio acido- base, vómitos, naúseas, alergia por hipersensibilidad, la cual se presenta sobretodo con la aspirina incluso a dosis pequeñas en cantidades de 300 mg provocando urticaria, edema angioneurotico y crisis de asma.5,9
La intoxicación por salicilatos debe ser tratada inmediatamente, caso contrario puede ser mortal, en caso de sospecha de intoxicación debe suspenderse el uso del salicilato inmediatamente, se puede realizar inducción de la emesis y lavado gástrico.5,9.
NOTAS
Univ. Tercer Año Facultad de Odontología UMSA
BIBLIOGRAFIA
1. Giancio S.B, Bourgaul. P.; Farmacología Clínica para Odontólogos; Analgésicos; Ed. "El Manual Moderno". 3ra edición; México D.F. Santa Fe 1990:5; 91-103. [ Links ]
2. Velásquez J; Farmacología Básica y Clínica; Fármacos antinflamatorias no esteroideos y otros analgésicos y antipiréticos; Editorial "Médica Panamericana S.A" 17ta edición. España. 2005:31; 519-522. [ Links ]
3. Rang H.P; Farmacología; Mediadores químicos; Editorial "Churchill Livinstone". USA.1995:12;303-314 [ Links ]
4. Hardman J., Limbird L.; Goodman y Gilman Las Bases Farmacológicas de la Terapéutica; "LitografiaIngranex". 10ma edición México 2005: (1); 27; 630-638.
5. Litter M.; Compendio de Farmacología; farmacología de la inflamación. Antinflamatorio no esteroides analgésicos antipiréticos. Antinflamatorios específicos. "El Ateneo". 4ta edición Buenos Aires.Argentina. 1992: 43; 608, 613-621. [ Links ]
6. Flores J.; Farmacología Humana; Fármacos analgésicos, antitérmicos, y antinflamatorios no esteroideos. Antriartricos; Editorial Elsevier Doyma. 4ta edición; Barcelona España. 2003:22; 385-390. [ Links ]
7. Katzung B.; Farmacología Básica y Clínica; Antinflamatorios no esteroideos. "El Manual Moderno". 10ma edición México -1999; 9: 592-598. [ Links ]
8. Neidle E.A Kroecer D.C Farmacología y Terapéutica Odontológicas Nueva Editorial Interamericana 1ra edición. México D.F-1984:10; 334-336 [ Links ]
9. Calderón O.; Boletín Informativo de Medicamentos. CIDME; No 30; enero La Paz- Bolivia; 1997:13,5-6 [ Links ]
10. Vademécum Especialidades Farmacéuticas Editorial "Campos Iris SRL" 10ma edición. La Paz - Bolivia .2010:3, 10,704. [ Links ]^rND^sGutiérrez Acosta^nAlex^rND^sGutiérrez Acosta^nAlex^rND^sGutiérrez Acosta^nAlex
ARTICULO
DERIVADOS ARILACETICOS O FENILACETICOS Y OXICAMS
Gutiérrez Acosta Alex 1
RESUMEN
Los derivados arilacéticos, fenilacéticos y oxicams son antiinflamatorios no esteroideos, (AINES) cuya función principal es inhibir la enzima ciclooxigenasa 2, sustancia fundamental en la formación de la prostaglandina durante el proceso de inflamación; y la enzima ciclooxigenasa 1, la cual está relacionada a la formación de la prostaglandina para la protección de la mucosa gástrica.
Algunos de estos antiinflamatorios poseen una actividad selectiva, ya sea por la ciclooxigenasa 1 o 2, lo cual significa que sus efectos adversos son mucho menores que los AINEs sin actividad selectiva.
Por otro lado, los antiinflamatorios con actividad selectiva pueden desembocar en otros efectos no deseados tales como: complicaciones cardiovasculares, que pueden presentarse incluso hasta un año después de haber concluido con el tratamiento. Tal es el caso del Rofenoxib, Celecoxib, Etoricoxib, Parecoxib y Valdecoxib, fármacos que fueron retirados del mercado por sus efectos colaterales, no obstante, este problema puede ser causado también por AINES sin actividad selectiva, como el Naproxeno. Es por eso que todos estos riesgos o complicaciones dependen únicamente del tiempo de tratamiento y la dosis a ser recetada para el paciente.
PALABRAS CLAVE
Prostaglandinas. Enzima ciclooxigenasa. Derivados Arilaceticos. Derivados de oxicams.
INTRODUCCION
Desde tiempos inmemorables, las diferentes civilizaciones humanas trataron de aliviar el dolor de muchas y diferentes maneras, los fenicios usaban la corteza del sauce y plantas pertenecientes a su familia. A mediados del siglo XVIII el reverendo Edmund Stone describió en una carta, el éxito obtenido con la corteza de dicho árbol para curar la fiebre intermitente. Tiempo después se descubrió que el ingrediente activo de la corteza de éste árbol era un glucósido amargo llamado salicilina, que por hidrolisis producía glucosa y alcohol salicílico. La salicilina fue aislada por primera vez en forma pura por Leroux en 1989, quien por su parte, también demostró sus propiedades antipiréticas. Más tarde se obtuvo el acido salicílico luego de escrupulosa manipulación química.
En 1875 el acido salicílico fue usado por primera vez para el tratamiento de la fiebre reumática y posteriormente para el tratamiento de la gota. Más tarde en 1899 Dresser introdujo éste compuesto al mercado farmacéutico como acido acetil salicílico o Aspirina, con propiedades antiinflamatorias. Ese fue el comienzo para un grupo de analgésicos antiinflamatorios no esteroideos y hasta el final del siglo XIX se comenzaron a descubrir otros grupos de fármacos, que fueron evolucionando muy rápidamente, como los derivados arilacéticos y los oxicams.
DEFINICION DEAINES
Los antiinflamatorios no esteroideos o AINEs son fármacos con efecto antiinflamatorio, analgésico y antipirético por lo que reducen los síntomas de la inflamación, alivian el dolor y la fiebre. El término no-esteroideo se refiere a que los efectos clínicos son similares a los de los corticoides pero no las acompañan las consecuencias secundarias que caracterizan a estos. Como analgésicos se caracterizan por no pertenecer a la clase de los narcóticos y actúan bloqueando la síntesis de prostaglandinas.
MECANISMO DE ACCION
Los fármacos antipiréticos antiinflamatorios no esteroideos son agentes terapéuticos que se usan para tratar la inflamación y el dolor, mediante la inhibición de las prostaglandinas impidiendo la acción de la enzima ciclooxigenasa, estas drogas comparten acciones farmacéuticas muy semejantes como:
1. Acción analgésica: Cuando se usan como analgésicos, éstas drogas suelen ser efectivas solo contra el dolor de intensidad baja o moderada, aunque sus efectos máximos son mucho menores, carecen de las acciones indeseadas sobre el sistema nervioso central que son frecuentes con los opiáceos, incluyendo depresión respiratoria y el desarrollo de dependencia física. Los AINES no cambian la percepción de las modalidades sensitivas, excepto la del dolor, éste puede ser post quirúrgico crónico o el que surge a causa de la inflamación y es muy bien controlado con esos agentes, mientras que no suelen aliviar el dolor que se origina en una víscera hueca.
2. Acción antipirética: Como antipiréticos los AINEs reducen la temperatura corporal en los estadios febriles. Aunque todas estas drogas son antipiréticas y analgésicas, algunas no son apropiadas para uso de rutina o prolongado debido a su toxicidad, como la fenilbutazona, derivado pirazolonico, que puede conducira anemia aplásica.
3. Acción antiinflamatoria: Estas drogas encuentran su principal aplicación clínica como agentes antiinflamatorios en el tratamiento de los trastornos músculo - esquelético, como artritis reumatoidea, osteoartritis y espondilitis anquilosante. En general, solo brindan alivio sintomático del dolor y la inflamación asociada con la enfermedad, reduciendo el progreso de la agresión patológica a los tejidos durante episodios graves.
EFECTOS ADVERSOS
Además de compartir muchas actividades terapéuticas, éstas drogas tienen en común varios efectos indeseables. Entre ellos, el más frecuente es la propensión a inducir alteraciones ulcerosas gástricas e intestinales con tendencia variables de acuerdo al tiempo de uso y dosis, que se acompañan a veces por anemia como resultado de la pérdida sanguínea. El daño gástrico producido por estos agentes puede deberse a no menos de dos mecanismos diferentes.
Aunque la irritación local por su administración oral permita la retrodifusión de ácido hacia la mucosa gástrica e induce daño tisular, también la vía parenteral puede ocasionar daño y sangrado. Esto parece estar correlacionado con la inhibición de la biosíntesis de prostaglandinas gástricas, en especial el PGE y PGI2 que inhiben la secreción ácido gástrica por el estómago y promueven la secreción de mucus citoprotector en el intestino; la inhibición de su síntesis puede hacer al estomago más susceptible al daño.
Otros efectos colaterales de estas drogas que tal vez dependan del bloqueo de la síntesis de prostaglandinas endógenas incluyen alteraciones en la formación plaquetaria, prolongación de gestación o trabajo de parto espontáneo y cambios en la función renal.
La función plaquetaria parece perturbarse por que estas drogas evitan la formación de tromboxano A2 por las plaquetas, que es un poderoso agente agregante. Esta acción es responsable de la tendencia de estas drogas de aumentar el tiempo de sangría. Como ya se mencionará, estos fármacos son inhibidores, particularmente efectivos, de la función plaquetaria, este "efecto colateral" ha sido explicado en el tratamiento profiláctico de los trastornos trombo embólicos. La prolongación de la gestación por AINEs se ha demostrado en animales de experimentación y en mujeres.
Las prostaglandinas de las series E y F son agentes útero trópicos potentes y el útero aumenta su biosíntesis en forma notable en las horas que preceden al parto. Por ello se ha teorizado que las prostaglandinas tienen un papel importante en la iniciación y progresión del trabajo del parto y el parto mismo.
Estos AINEs tienen poco efecto sobre la función renal de los seres humanos normales, tal vez porque la producción de prostaglandinas vasodilatadoras solo tiene un papel menor en los individuos con carga de sodio normal, éstas drogas disminuyen el flujo sanguíneo renal y el índice de filtración glomerular en los pacientes con insuficiencia cardiaca congestiva, cirrosis hepática con ascitis o nefropatía crónica o en aquellos que están hipovolemicos por cualquier razón en estas circunstancias puede precipitarse una insuficiencia renal aguda.
En todos estos cuadros, la perfusión renal es más dependiente de las prostaglandinas que causan vasodilatación y que pueden oponerse a las influencias vasoconstrictoras de la noradrenalina y la angiotensina II que resultan de la acción de los reflejos presores.
Además de los efectos hemodinámicos en el riñón, también promueven la retención de sal y agua mediante la reducción de la inhibición inducida por las prostaglandinas sobre la reabsorción de cloruros y la acción de hormona antidiuretica. Esto puede causar edema en algunos pacientes que son tratados con estas drogas; también puede reducir la efectividad de los regímenes antihipertensivos. Estos agentes promueven hiperpotasemia mediante varios mecanismos, incluyendo el aumento de la reabsorción de K+, como resultado de la disminución de la disponibilidad de sodio en los túbulos distales y de supresión de la secreción de renina inducida por las prostaglandinas.
El último efecto puede ser responsable en parte de la utilidad de estas drogas en el tratamiento del síndrome de Bartter, que se caracteriza por hipopotasemia, hiper-reninemia, hiperaldosterismo, hiperplasia yuxtaglomerular, normotension y resistencia al efectopresor de la angiotensina II. La producción excesiva de las prostaglandinas renales puede tener un papel importante en la patogenia de este síndrome.
A continuación se presentan tablas comparativas que describen a los derivados arilaceticos y oxicams.
DERIVADOS ARILACETICOS
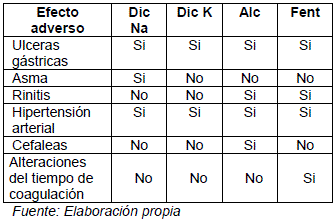
Este grupo de derivados arilacéticos o fenilacéticosen especial puede producir efectos no deseados como toxicidad renal, toxicidad hematológica y reacciones de hipersensibilidad. Los fármacos de este grupo son:

TABLA COMPARATIVA EN TRATAMIENTO DE ENFERMEDADES
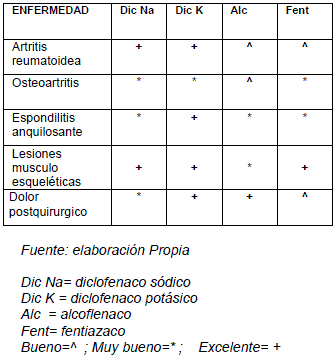
TABLA DE EFECTOS ADVERSOS DE DERIVADOS ARILACÉTICOS
DERIVADOS DE LOS OXICAMS
Por otro lado los oxicams son un grupo de AINEs quienes poseen un tiempo de vida prolongado, y de la misma forma su eliminación se produce lentamente.
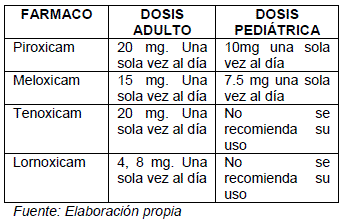
TABLA DE EFECTOS ADVERSOS DE DERIVADOS OXICAMS
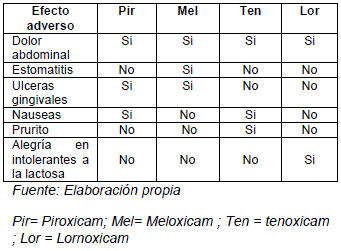
NOTAS
1 Univ. Tercer Año Facultad de Odontología UMSA
BIBLIOGRAFIA
1. Baños Díez J. y Farré Albaladejo M. Principios de farmacología clínica, 1ra edición. Editorial Masson, España -Barcelona, 2002:15-17, 30, 31. [ Links ]
2. Flores J. Farmacología humana. 4ta edición. Editorial Masson. España -Barcelona, 2005: 58 -61 [ Links ]
3. Goodman y Gilman. Bases de la terapéutica farmacológica. 11ra edición. Editorial MgGraw Hill. USA. 2006:261 -264 [ Links ]
4. Katzung B. Farmacología básica y clínica, 10ma edición, McGraw - Hill Lange. USA - San Francisco, 2006: 177-180 [ Links ]
5. Litter. Farmacología experimental y clínica. 7ma edición. Editorial Ateneo. Argentina - Buenos Aires, 1986: 89 -92 [ Links ]
6. Loza E. Aines en la práctica clínica: lo que hay que saber. Madrid, España. 2011. http://www.msc.es/biblioPublic/publicaciones/recursos_propios/infMedic/docs/vol35n3AINEs.pdf. Fecha de Acceso: 19 de septiembre 2012. [ Links ]
7. Mendoza Patiño N. Farmacología médica. 1ra edición. Editorial Panamericana. México DF. 2008; 55-57 [ Links ]
8. Netter F.H. , Farmacologia ilustrada, 1ra edición, editorial Icon Learning System, USA- New Jersey, 2005: 90 -92 [ Links ]
9. Cort R., Ponsoda X., Jover R.. Toxicidad del diclofenaco en los hepatocitos: el papel del metabolismo de la droga en la toxicidad de las células. 1999. Disponible en: http://www.ncbi.nlm.nih.gov/pubmed/986275 Fecha de acceso : 20 de septiembre 2012. [ Links ]
10. Yasushiro M., Haruka S., Toshiharu H.. Requerimientos estructurales para la hepatoxicidad de antinflamatorios no esteoideos: pruebas en hepatocitos de ratón. 1998. Visto en el link: http://jpet.aspetjournals.org/content/287/1/208.long Fecha de acceso 20 de septiembre 2012. [ Links ]^rND^sBustamante C^nGladys^rND^sGutierrez Aguilar^nHiver Mauricio^rND^sBustamante C^nGladys^rND^sGutierrez Aguilar^nHiver Mauricio^rND^sBustamante C^nGladys^rND^sGutierrez Aguilar^nHiver Mauricio
ARTICULO
AINES DERIVADOS PIRROLACETICOS, PIRAZOLONICOS, FENAMATOS
Mgs. Dra. Bustamante C. Gladys 1
Gutierrez Aguilar Hiver Mauricio 2
RESUMEN
Uno de los puntos focales del uso de los aniinflamatorios no esteroideos, se relaciona al control del dolor en su fase aguda o crónica, es por ello que se han desarrollados grandes esfuerzos para lograr elaborar medicamentos que controlen este desagradable síntoma en forma rápida, eficaz y con la menor cantidad de efectos colaterales posibles.
Es en este sentido, que luego del descubrimiento de la salicilina, que dio origen a la aspirina (ASA) , numerosos compuestos han sido experimentados con la intención de mejorar la respuesta de este producto, de este modo es que hace pocos años se dio lugar a la aparición de derivados del ácido acético, con potencia analgésica mayor al proporcionado por el ácido acetil salícilico, como es el caso del Ketorolaco, útil para procesos dolorosos moderados a severos con potencia similar a la morfina, u el metamizol, útil para dolor moderado principalmente de tipo cólico con mayor efecto antitérmico que otros AINES, o la fenilbutazona, retirada ya del mercado por sus grandes efectos colaterales.
El presente artículo tiene como fin realizar un acercamiento sobre algunos de estos fármacos.
PALABRAS CLAVE
Ketorolaco. Metamizol. Clonixinato de Lisina. Fenilbutazona
INTRODUCCION
El hablar de la farmacología del dolor lleva a la comprensión y análisis de varios compuestos, que tienen como fin el manejo de uno de los síntomas más molestos en el área de la salud, y que en ocasiones lleva a acciones terapéuticas de mayor severidad ante la falta de su control.
La capacidad del profesional radica en identificar el estado de ansiedad que este síntoma provoca en los pacientes, y observar la misma durante la terapia a instaurar, con el fin de mejorar la calidad del paciente, reducir sus niveles de estrés, reducir el tiempo de tratamiento y evitar técnicas invasivas para controlar dicho síntoma.1
El control farmacológico del dolor se realiza con diferentes medicamentos que corresponden inicialmente al grupo de los AINES, que en caso de fracasar en su respuesta, permitirán el uso de otro tipo de analgesia, con corticoides, narcóticos, anestésicos, etc., debiendo de acuerdo al caso, iniciar la analgesia con AINES, en función al caso, intensidad del dolor, tiempo de respuesta esperada, etc.
ANTIINFLAMATORIOS NO ESTEROIDEOS
Los antiinflamatorios no esteroideos incluyen un amplio contingente de medicamentos, los cuales se organizan de acuerdo al grupo de origen. De esta manera los grupos a ser estudiados en éste artículo son derivados:2-4,7
a) Pirrolacéticos: donde se encuentran el ketorolaco y la tolmetamina.
b) Pirazolonicos: o pirazolindindionicos como la antipirina, aminopirina (retiradas por mutagenas y carcinogénicas), dipirona fenilbutazona (retirada por toxicidad hematológica), febrapzona, metamizol, nifenazona, ixipozona, pinazona y propifenazona, oxifenbutazona, gamacetofenilbutazona, clofenazona, bumidazona, azapropazona
c) Fenamatos: como el ácido, mefenámico, flufenámico nifumínico. meclofenámico, etofenamato.5,9
Muchos de los cuales han sido retirados del mercado por sus efectos colaterales, como en el caso de la fenilbutazona, aminopirina y antipirina.3,6,9
MECANISMO DE ACCION
El mecanismo de acción de los AINES se orienta hacia el efecto analgésico, antiinflamatorio, anti térmico y anti agregante plaquetario, a partir de los cuales, la diferencia de potencia de acción varía de acuerdo a la familia de origen del medicamento, existiendo variaciones independientes sobre los efectos colaterales, que pese a tener una similitud, generan diferencias en base al origen del medicamento, dosis y vía de administración.
En tal sentido se puede mencionar que los AINES son un grupo heterogéneo de compuestos que no se relacionan químicamente, siendo la mayor parte de ellos, ácidos orgánicos, que comparten acciones y efectos colaterales, mientras que aquellos que corresponden a los ácidos inorgánicos (fenilbutazona, propifenazona, metamizol) tienen la misma similitud.
Es de allí que el primer efecto sobre el cual trabajan los AINES, se relaciona a la limitación del dolor inhibiendo la enzima ciclooxigenasa (COX,), la cual tiene dos isoenzimas la 1 y 2 codificadas por genes diferentes. La isoenzima COX1 interviene en la formación de prostaglandina I con acción antitrombogénica y protectora de la mucosa gástrica, mientras que la COX2 se encuentra solamente en tejidos con inflamación, aumentando sus niveles por influencia de los fibroblastos, citoquinas, etc. 4,6
Por lo tanto los AINES inhibirán a la formación de prostaglandinas a partir del ácido araquidónico de diferentes tipos celulares, compitiendo por el mismo durante la respuesta inflamatoria, acoplándose al sitio activo de los canales enzimáticos, reduciendo el dolor y la inflamación.5 (5)
Los AINES en forma general inhiben el Tromboxano A2 4,5, reduciendo la agregación plaquetaria, esta inhibición puede ser reversible.
Mientras que la acción antipirética se relaciona a
DERIVADOS PIRROLACETICOS
En el grupo de los derivados pirrolacéticos se tiene al ketorolaco como medicamento representativo, es así que este medicamento denominado también como ketorolaco trometamia es un antiinflamatorio no esteroideo de la familia de los derivados heterocíclicos del ácido acético, derivado del ácido arilpropiónico, que tiene un alto nivel analgésico inhibiendo débilmente las prostaglandinas, comparándose su acción al de los analgésicos opiáceos. Su acción sobre la ciclooxigenasa es dependiente de la dosis, compite con el sustrato, teniendo una biodisponibilidad con el consumo oral del 90% y muscular del 100%.
El consumo de los alimentos retrasa la absorción digestiva, sin modificar su potencia, difunde poco por la barrera hematoencefálica y mamaria, pero pasa con facilidad a la placenta.
Su acción analgésica con administración por vía intramuscular se inicia a los 10 min, y administrado por vía oral comienza a los 30-60 min, alcanzando su pico máximo de acción a las 1,2-3 y 1,5-4 horas, permaneciendo con niveles estables por 6 a 8 horas. Es el primer medicamento analgésico para uso endovenoso e intranasal.3,7,8
Una vez administrado, se une a las proteínas plasmáticas fijándose en un 99% y se metaboliza en el hígado, dando metabolitos sin acción analgésica ni antiinflamatoria útil, eliminádose por orina un 91% y el 3% restante se elimina por vía biliar, durante las 4-6 horas posteriores a su ingesta.3
Sus efectos adversos son similares al de los demás AINES, siendo importantes de mencionar: las hemorragias gastrointestinales, broncoespasmo, síndrome de Stevens Johnson, síndrome de Lyell, edema angioneurótico, etc.
Su uso está contraindicado en hipersensibilidad a los AINES, anemia aplásica y deficiencia de glucosa 6 fosfatodeshidrogenasa, debiéndose tener precaución en pacientes con hipertensión arterial sistémica, insuficiencia renal o hepática, discrasias sanguíneas, entre otras.3,7
PIRAZOLONAS
Las pirazolonas son inhibidores competitivos de la ciclooxigenasa y al igual que los demás AINES, tienen los efectos, analgésicos, antipiréticos y antiinflamatorios.
El metamizol es el agente de mayor uso dentro de este grupo, y representa la mayor parte de acciones correspondientes a los integrantes de esta familia.
Es de esta forma remarcable la acción analgésica del metamizol y la propifenazona, los cuales actúan con potencia analgésica en dolor moderado, con acción similar a la aspirina, siendo menos lesivo para la mucosa gástrica que ésta última, además posee acción relajante sobre la musculatura lisa, por lo que se indica en dolor de tipo cólico.6,7
Es de hacer notar que su acción antiinflamatoria es de muy bajo poder al igual que su acción antipirética.5-7
Al igual que el ketorolaco, se absorbe bien por vía oral, adhiriéndose a las proteínas plasmáticas en un 90%. Su metabolismo es hepático en un 90% y su excreción se realiza por vía renal.
La administración del metamizol debe hacerse con precaución, ya que a diferencia de otros AINES provoca agranulocitosis con mayor facilidad, y de igual forma que otros antiinflamatorios no esteroideos puede inducir rush, urticaria, angioedema, naúseas, vómitos. La posibilidad de provocar oliguria, anuria, nefritis intersticial, y anemia hemolítica idiosincrática la hace diferente a los demás medicamentos de los grupos en mención.
Su aplicación por vía endovenosa puede provocar irritación local, y por vía endovenosa puede llevar al paciente a cuadros de hipotensión arterial, asociados a naúseas, rubor facial, sensación de calor y palpitaciones.
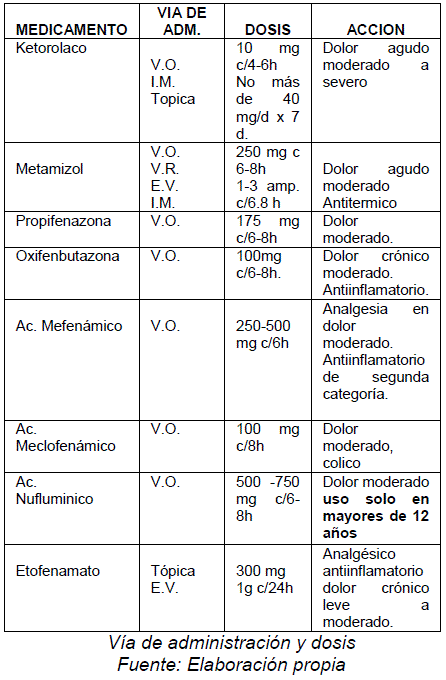
FENAMATOS
Son aquellos medicametnos derivados del ácido arilantranílico, que son utilizados desde 1950, sin mayores diferencias en relación a la aspirina.
El medicamento representativo del grupo y aquel que se utiliza con más frecuencia en el comercio, es el ácido mefenámico, el cual sirve principalmente para disminución del dolor en procesos artropáticos y dismenorrea, éste medicamento es el único fenamato que presenta acciones centrales y periféricas para producir analgesia, teniendo potencia antiinflamatoria menor a la fenilbutazona (50%), mientras que el ácido flufenámico es 50% más potente que la primera droga.
Este grupo de medicamentos se administran por vía oral, alcanzando una concentración máxima entre 30 y 120 min., con una vida media de 2 a 4 h. eliminándose por orina en forma de 3 hidroxilatos y/o carboxilatos, su eliminación por heces es de solo el 30% de la concentración plasmática.
Los efectos colaterales son similares a los otros AINES, siendo sobresalientes, la esteatorrea, diarrea, anemia hemolítica, por lo que se recomienda no utilizarlos en pacientes con antecedentes de estos cuadros.7,8
En este grupo es remarcable un nuevo medicamento, el etofenamato que tiene aplicación percutánea, que tiene acción local y también sistémica, disminuyendo la inflación zonal, con menor toxicidad que el ácido flufenámico, es también notable la potencia antiinflamatoria ya que a diferencia de algunos otros AINES, incluyen la inhibición de la bradiquinina y de la serotonina a través de un efecto estabilizador de membrana. Su vida media es de 1,6 horas, alcanzando su concentración plasmática máxima a las 2h.9
NOTAS
1 Médico Internista. Docente Emérito Semiología General UMSA. Mg.Sc. Psicopedagogía y Educación Superior.
2 Univ. Tercer Año Facultad de Odontología UMSA
BIBLIOGRAFIA
1. Control del dolor Universidad de Valparaiso URL disponible en : http://es.scribd.com/doc/62324544/5/FENAMATOS-DERIVADOS-DEL-AC-ANTRANILICO Fecha de acceso: 1 de octubre del 2012 [ Links ]
2. Ramírez Guerrero A. Burlch-Bonechi J. Dolor agudo postoperatorio, su frecuencia y manejo. Rev. Mex. Anest: 1992:77;439-445 [ Links ]
3. Argueta López R. Propiedades farmacológicas básicas del ketorolaco trometamina 10 mg. Oral en analgesia perioperatoria para cirugía dental. Congreso Virtual de Anestesiologia México. URL disponible en: http://www.slideshare.net/ravetmx13/propiedades-farmacologicas-basicas-del-ketorolaco-trometamina-10-mg-oral-en-analgesia-perioperatoria-para-ciruga-dental Fecha de acceso 1 de octubre del 2012 [ Links ]
4. Delgado de Molina P. AINEs preferentemente inhibidores de la COX2, complicaciones de los COX2 URL disponible en: http://revista.sedolor.es/pdf/2000_09_03.pdf Fecha de acceso 1 de octubre del 2012. [ Links ]
5. Farmacología de los analgésicos no opiáceos AINEs. Capitulo 6. URL disponible en: http://www.catedradeldolor.com/PDFs/Cursos/Tema%206.pdf. Fecha de acceso 1 de octubre del 2012 [ Links ]
6. Criterios de selección de un analgésico no esteroideo URL disponible en: http://2011.elmedicointeractivo.com/farmacia/temas/tema5-6/criterios5.htm?botsearch [ Links ]
7. Valsecia M. Analgésicos antipiréticos y antiinflamatorios no esteroides (AINEs) cap. 7. URL disponible en: http://med.unne.edu.ar/catedras/farmacologia/temas_farma/volumen4/cap7_aines.pdf. Fecha de acceso 1-10-2012 [ Links ]
8. Farmacología de los analgésicos no opioides. 1-18 URL disponible en: http://www.plandolor.grupoaran.com/pdfs/Uni3_1.pdf. Fecha de acceso 1 de octubre del 2012. [ Links ]
9. Carretero M. Medicamentos de Vanguardia. Etofenamato. URL disponible. http://apps.elsevier.es/watermark/ctl_servlet?_f=10&pident_articulo=10022249&pident_usuario=0&pident_revista=4&fichero=4v20n03a10022249pdf001.pdf&ty=110&accion=L&origen=doymafarma&web=www.doymafarma.com&lan=es. Fecha de acceso: 1 de octubre del 2012. [ Links ]^rND^sRamírez Guerrero^nA^rND^sBurlch-Bonechi^nJ^rND^sGutiérrez Carrillo^nCristhian^rND^sValencia Callejas^nSamira^rND^sGutiérrez Carrillo^nCristhian^rND^sValencia Callejas^nSamira^rND^sGutiérrez Carrillo^nCristhian^rND^sValencia Callejas^nSamira
ARTICULO
DERIVADOS DEL ACIDO PROPIONICO Y DERIVADOS DEL PARA-AMINOFENOL
Gutiérrez Carrillo Cristhian 1
Valencia Callejas Samira 2
RESUMEN
Los derivados del ácido propiónico y del para-aminofenol son un conjunto de drogas que pertenecen a la familia de los AINEs (antiinflamatorios no esteroideos), los cuales poseen propiedades antiinflamatorias, analgésicas, antipiréticas e inhibitorias de la agregación plaquetaria.
Estos fármacos están formados por diferentes compuestos químicos, es por eso que pueden lograr tener cada una de sus propiedades características en mayor o menor grado y se suministran en una terapia correspondiente según el caso lo amerite.
La comercialización de estos productos puede ser de venta libre o según su prescripción médica, ya que éstos fármacos son adquiridos y utilizados comúnmente para calmar dolencias que van desde una alteración menor en el organismo como un trauma o para el tratamiento de una enfermedad más compleja como la artritis reumatoidea, por tanto es responsabilidad tanto del consumidor como también del médico profesional conocer cómo actúan estas drogas en nuestro organismo, qué beneficios y que adversidades tienen, ya que cada integrante de la gran familia de los AINEs, obtienen diferentes tipos de reacciones en el organismo, que van relacionadas con la cantidad o dosis ingerida de la droga.
PALABRAS CLAVE
AINES. Prostaglandinas.
INTRODUCCION
Los derivados del ácido propiónico y los derivados del ácido para-aminofenol son fármacos eficaces frente al dolor leve y moderado, poseen propiedades analgésicas, anti inflamatorias y antipiréticas como también están encargados de inhibir la agregación plaquetaria.
Las prostaglandinas como el tromboxano son formadas a partir de los fosfolípidos presentes en la membrana celular como ser el ácido araquidónico, que al sufrir un proceso de transformación llega a convertirse en ciclo oxigenasa (COX), las cuales pueden diferenciarse en (COX - 1 y COX - 2), ambas cumplen funciones completamente diferentes:
1. La COX1 cumple una función exclusivamente interna, teniendo como funciones esenciales la de proteger a la mucosa gástrica formando mucus gástrico y disminuyendo la formación de ácido gástrico para así mantener íntegra la homeostasis de la misma, es decirse encuentra ocupada por los ácidos gástricos sin producirse en ésta ninguna lesión como las úlceras gástricas (las cuales pueden ser provocadas por los AINEs por su cualidad inhibitoria de las prostaglandinas) y segundo una función igual de importante que se halla relacionada con mantener la hemostasia del organismo, ya que si se presenta una lesión que desencadenaría una hemorragia, ésta COX tendrá como misión la agregación plaquetaria en la lesión para formar el coagulo y de esta manera no alterar la circulación sanguínea.1,2
2. En cambio la COX2 está relacionada con el factor externo, es decir la inflamación el dolor y la temperatura; los AINES actuarán directamente inhibiendo éstas reacciones y consiguiendo la disminución de todas estas.
Particularmente aquellos que actúan como antiinflamatorios, se desenvuelven produciendo una disminución en la formación de prostaglandinas en los tejidos periféricos, por tanto no permiten el proceso inflamatorio, inhibiendo la vasodilatación y el edema, es decir que si bien sus efectos son periféricos, actuará en sí a nivel del sistema nervioso central.3,4
Su acción analgésica se da por la disminución de la sensibilidad de las terminaciones nerviosas nociceptoras ya que tales terminaciones nerviosas están encargadas de recibir la sensación dolorosa y transmitirla a las neuronas sensitivas ubicadas en SNC para posteriormente dirigirse al cerebro. También actúan como antipiréticos, porque la temperatura será regulada en caso de presentarse alterada y dicha regulación será mediada por el hipotálamo por ser éste el encargado de regular directamente la COX2, es decir el AINE irá a actuar a nivel hipotalámico en éste único caso y no a nivel del SNC.1
DERIVADOS DEL ACIDO PROPIONICO
Los derivados del ácido fenil propiónico o ácido propiónico, son AINES que presentan variantes en su estructura química y sus propiedades farmacocinéticas pero no diferirán en cuanto a su acción farmacológica ni sus reacciones adversas, es decir son semejantes en cuanto a sus características farmacológicas. Entre éstas las propiedades características que poseen los AINES están las propiedades antiinflamatorias, antipiréticas, analgésicas y antiagregante plaquetaria, que los hace muy parecidos a la aspirina pero presentan una incidencia menor en cuanto a los efectos adversos.
Todos se absorben por vía oral, pero los alimentos retrasan su absorción y por vía rectal , que es mucho más lenta e irregular. Se difunden bien alcanzando concentraciones entre 50 y 70% en el plasma. Pasan por la placenta y se encuentra en una cantidad mínima en la leche materna. Sus reacciones adversas son parecidas a los demás AINES, se caracterizan por tener menor incidencia en la mucosa gastrointestinal.5,8
1. Ibuprofeno: El ibuprofeno, fue el primero de este grupo y su introducción fue en 1969 para utilizarse como una alternativa del AAS ya que es más tolerable.1
Su efecto es benéfico en el tratamiento de la artritis reumatoide y en dosis más altas para la artrosis, ya que puede evidenciarse la reducción tanto de la tumefacción articular como del dolor y de la duración de la rigidez matinal, progreso de la capacidad funcional manifestada por aumento de la fuerza de presión y el retardo de la aparición de fatiga. También está indicado en el tratamiento de la dismenorrea primaria, alivio del dolor que va de leve a moderada y como antipirético.
Su absorción es rápida posterior a su administración oral, se metaboliza rápidamente y su excreción es por la orina; su vida media alcanza 2 horas aproximadamente.
En dosis más bajas se lo utiliza como analgésico y antipirético (de 200 a 400 mg cada 4 a 6 horas). A dosis más elevadas se maneja como
antirreumático, puede producir los mismos efectos que los demás AINES (en adultos la dosis oral es 1.200 a 3.200mg/día dividida en 3-4 tomas y en niños será de 20-40 mg/Kg/día en 3-4 tomas).3,7
Los efectos adversos de este grupo son leves lo que le confiere relativo éxito, los efectos colaterales mas comunes son molestias gástricas por irritación de ésta, nauseas, vómitos, problemas hemorrágicos, erupciones cutáneas, edemas periféricos. En el sistema nervioso central, se presentacefalea, mareos, visión borrosa o disminuida, escotomas, ansiedad y depresión; en caso de presentar estas alteraciones (molestias) la ingesta de esta droga debe suspenderse.
Su administración está contraindicada en individuos sensibles a la droga o con pólipos nasales, angioedema y reactividad broncospástica a la aspirina u otro AINE. Se han reportado casos de úlcera péptica y sangrado gastrointestinal, por tanto debe monitorearse su administración bajo estrecha vigilancia a pacientes con antecedentes de enfermedad del tracto gastrointestinal superior.
2. Naproxeno: Es utilizado para calmar el dolor que va de leve a moderado como también para la sensibilidad e inflamación, cefalea, dolor menstrual y dolores musculares, para este tipo de padecimientos no es necesaria una prescripción médica pero en caso de enfermedades como las artropatíasse utilizan dosis únicamente prescritas por el profesional en salud.
Este fármaco es asimilado y procesado en el hígado, y se elimina casi completamente por vía urinaria.
En adultos la dosis es de 500 mg al principio de la terapia como dosis de ataque, seguido de 250 mg cada 6 a 8 horas y como antirreumático se empleará 250-500mg cada 12 horas. Como antirreumático en niños se utilizará 10 mg /Kg /día dividida en dos tomas.3,7
Las reacciones adversas se pueden producir alergias, a nivel gastrointestinal, puede producir vómitos, nauseas, hemorragia gástrica y entre sus efectos centrales es posible encontrar adormecimiento, cefalea, mareos, cansancio corporal y desánimo llegando a un estado de depresión.
3. Fenoprofeno: Se utiliza en tratamientos para aquellas enfermedades que presenten síntomas de artritis, y también es utilizada como antipirético, analgésico y antiinflamatorio.
Este fármaco es asimilado y procesado a través del hígado y es eliminado casi por completo por la orina; su tiempo de vida en el organismo es de una 45 a 180 min. aproximadamente.
Se utiliza como analgésico si el dolor es leve o moderado (se administrara 200 mg cada 4-6 horas) y como antirreumático (entre 300 a 600 mg cada 6 a 8 horas).En los infantes no hay una dosis recomendada.3,4
Las reacciones adversas de éste, se encuentran a nivel gastrointestinal, además de debilidad muscular, cefalea, visión borrosa y somnolencia, y está contraindicado en personas alérgicas a éste producto y en individuos con alteraciones gástricas.
4. Ketoprofeno: Es utilizado para aliviar el dolor leve a moderadoy en caso de artritis reumatoide, osteoartritis y dismenorrea.
Una vez ingerida ésta droga, es absorbida por el tracto gastrointestinal, para luego concentrarse en el plasma y llegar de esta forma hacia el hígado por el cual será metabolizado; la droga tendrá una vida media de aproximadamente 2 horas y finalmente ésta será eliminado por la orina.
Como analgésico, en dolor leve a moderado se administra en dosis de 25 a 50 mg cada 6 a 8 horas, como antirreumático al principio se dosificará 150 mg a 300 mg cada día de 3 a 4 dosis al día.3,4
Está contraindicado en personas que presenten una reacción alérgica después de usado el mismo. Las reacciones adversas están relacionadas con efectos gastrointestinales y renales ocasionando en el primero una dispepsia leve, mientras que en el segundo una falla renal aguda.
5. Flubiprofeno: Es utilizado en enfermedades agudas, para el tratamiento artritis reumatoide, osteoartritis, espondilitis anquilosante, bursitis aguda, gota aguda, dismenorrea y dolor musculo esquelético.
Se metaboliza en el hígado, su duración en el plasma sanguíneo es de 45 min. y su tiempo de vida dentro del organismo es de 5 a 8 horas, eliminándose por la orina.
Se utiliza éste como antiinflamatorio (50 mg cada 4 a 6 horas), y como antidismenorreico (50 mg cada 6 horas), como antirreumático en principio 200 a 300 mg de 2 a 4 dosis por día). 3,6
Tiene reacciones adversas en mujeres embarazadas, también está relacionado con la ulcera péptica y los trastornos de la coagulación.Está contraindicado en personas hipersensibles y alérgicas al producto.
6. Oxaprozina: Utilizado enel alivio del dolor leve a moderado, también actúa como antiinflamatorio y se lo utiliza en el tratamiento de la artritis reumatoide, artrosis y gota.
Es metabolizado por el hígado y excretado casi completamente por la orina.
Es administrado por vía oral en tratamiento de artritis reumatoide con una dosis de 1.200 mg/día (toma única), en el tratamiento de la artrosis 1.200 mg/día como dosis de ataque y de 600 a 1.200 mg como dosis de mantenimiento, recomendándose utilizar máximo 26 mg/kg, repartida en varias tomas.3
Sus reacciones adversas están relacionadas con efectos gastrointestinales produciendo vómitos, dolor abdominal y sus efectos centrales son adormecimiento, cefalea, mareos y cansancio corporal. Está contraindicado para aquellas personas con reacciones alérgicas, en mujeres embarazadas.
7. Indoprofeno: Medicamento utilizado en enfermedades como artritis reumatoide, osteoartritis y cáncer. Éste fármaco actualmente ya no es recomendado por su carcinogenicidad.
DERIVADOS PRINCIPALES DEL PARA-AMINOFENOL
El uso del para-aminofenol fue disminuyendo por sus efectos tóxicos ya que sus metabolitos formarán radicales altamente reactivos cancerígenos y también es el responsable de la producción de metahemoglobinemia. Presenta derivados como: el paracetamol o acetaminofeno (derivado acetilado del ácido para-aminofenol) el cual es el más utilizado; también presenta derivados que a su vez son drogas derivadas de la anilina o fenilamina, entre estos la fenacetina o cetofenitidina(derivado éter etílico de la antigua "acetanilida" la cual se encuentra en desuso por su toxicidad).
Estos derivados son potentes analgésicos y antipiréticos por su acción inhibitoria de la COX2, pero carecen de propiedades antiinflamatorias por la escasa inhibición de la síntesis de las prostaglandinas implicadas en la inflamación. Otra acción característica de este grupo es el de poseer una acción sedante en el organismo, por tanto en dosis elevadas podría producir el descenso de la presión arterial que conlleve a la depresión cardiaca. Se absorben por todas las vías y por su característica de ser una base muy débil se absorbe algo en el estómago y muy rápidamente en el intestino.1
1. Paracetamol: Es un fármaco muy eficaz como analgésico y antipirético, carece de propiedad antiinflamatoria y que es tolerado y seguro de usar en dosis terapéuticas, pero la sobredosis del mismo puede causar una lesión hepática.
Se absorbe por el intestino delgado y dependiendo del vaciado gástrico, se metabolizado por el hígado siendo su biotransformación rápida y su eliminación se da por la orina lo que le confiere a ésta una coloración roja pardusca. La vida media es de 2.5 horas. La absorción por via rectal es lenta.1
Posee una actividad sobre la COX1 y COX2, inhibiendo la síntesis de prostaglandinas.Se lo utiliza cuando la ASA está contraindicada y la dosis que es de (500 mg cada 4 a 6 horas), sin sobrepasar los 4 g diarios. Las dosis pediátricas son de 10 mg/Kg de peso repartidos entre 4-5 tomas.5
En dosis terapéuticas es tolerado por el organismo, la reacción adversa más importante es la intoxicación aguda en la cual puede presentarse necrosis hepática con aparición de náuseas, vómitos, ictericia, dolor hepático, orina oscura, oliguria y azoemia.
2. Fenacetina :Se la obtiene por reemplazo de un hidrógeno del grupo amino en la anilina por el radical acetilo "acetanilida" disminuye la toxicidad pero el compuesto aún es bastante tóxico ya que la acetanilida es un directo derivado acetilado del ácido para-aminofenol.7
Este fármaco se caracteriza por sus propiedades antipiréticas y analgésicas, como también por ser un inhibidor de las prostaglandinaspero carece de propiedades antiinflamatorias. Actualmente se utiliza poco debido a sus efectos adversos hepáticos, hematológicos y su nefrotoxicidad.La fenacetina puede causar metahemoglobulinemia sulfahemoglobilemia y anemia hemolítica y la administración a grandes dosis de mezclas que en su composición contengan fenacitina son relacionadas con el desarrollo de una necrosis papilar renal.4-10
NOTAS
1 Univ. Tercer Año Facultad de Odontología UMSA
2 Univ. Cuarto Año Facultad de Odontología UMSA. Redactora
BIBLIOGRAFIA
1. Rang y Dale. Farmacia. 7ma edición. España; Editorial Elsevier; 2008:227-2350. [ Links ]
2. Stockley. Interacciones Farmacológicas. 2da edición. Barcelona. Editorial Pharma Editores. 2006:520-526 [ Links ]
3. Bertram G. Katzung. Farmacología Básica y Clínica. 10ma edición. México; Editorial El Manual Moderno. 2007: 587-598. [ Links ]
4. Litter Ml. Compendio de Farmacología. 4ta edición. Venezuela: Editorial El Ateneo; 1988: 608-638. [ Links ]
5. Remington. Farmacia. 20ma edición. Buenos Aires-Argentina; Editorial Panamericana. 2003:1727-1736 [ Links ]
6. Tripathi. K.D. Farmacología en Odontología: Fundamentos. 1ra. Edición Buenos Aires -Argentina.: Editorial Médica Panamericana. 2008:335-345. [ Links ]
7. Flores J. Farmacología Humana. 4ta edición. Barcelona . Editorial Elsevier Masson.2003: 165-180. [ Links ]
8. Silva Reegiardo E. Manejo de los problemas pulpares en la dentición temporal. 1ra edición. México. Editorial Trillas. 1999:61-68. [ Links ]
9. Fernández Sánchez J. Manual de prácticas. 1ra edición. Madrid España: Editorial Amolca. 2006: 157-163. [ Links ]
10. Luis Isla J, Síntesis de la Fenacetina. del 2012.Paginas 3-17. Disponible: en http://es.scribd.com/doc/44732537/Farmaco-fenacetina. Fecha de acceso 28 de septiembre [ Links ]^rND^sEscalante Cuba^nNataly Carla^rND^sFernández Yujra^nLidia Eugenia^rND^sEscalante Cuba^nNataly Carla^rND^sFernández Yujra^nLidia Eugenia^rND^sEscalante Cuba^nNataly Carla^rND^sFernández Yujra^nLidia Eugenia
ARTICULO
ANALGESICOS OPIOIDES FENANTRENICOS
Escalante Cuba Nataly Carla 1
Fernández Yujra Lidia Eugenia 2
RESUMEN
Los analgésicos opioides fenantrénicos son fármacos derivados del opio ,que presentan propiedades analgésicas potentes al interactuar con receptores denominados mu (μ), kappa (k) y Delta (δ), siendo sus principales representantes la morfina y la codeína, el primero tiene su acción por antagonismo del receptor mu (μ) mayor que el resto de los receptores opioides, sin embrago los efectos colaterales que presenta como las naúseas, vómitos, depresión respiratoria, mareos, coma, miosis y farmacodependencia han hecho que su uso sea muy limitado. Esta droga se contraindica en pacientes con asma y con lesiones cerebrales.
Por su parte, la codeína con acción analgésica 10 veces menor al opio tiene poca afinidad por los receptores opioides, requiriendo combinarse con AINES para mejorar dicha acción, por lo que su uso se limita en dolor leve y moderado, además de la sedación de la tos y está contraindicado en personas con depresión respiratoria, asma e hipersensibilidad al medicamento, debiendo tomarse precauciones en el uso de pacientes embarazadas, insuficientes hepáticos o renales, ya que el uso prolongado podría llevar a farmacodependencia..
PALABRAS CLAVES
Morfina. Codeína. Metilmorfina. Alcaloides Naturales
INTRODUCCION
Los analgésicos opiodes son fármacos obtenidos de la amapola (Papaver somniferum) a partir de una sustancia lechosa líquida, que luego se deseca, obteniéndose un polvo con más de 20 alcaloides organizados en dos grupos: los derivados fenantrénicos y los benzilisoquinolinicos.1 Estos fármacos comparten propiedades semejantes uniéndose a receptores opiodes como las encefalinas, endorfinas y dinorfina, lo que les dan sus propiedades analgésicas e hipnóticas. Los representantes sobresalientes del grupo de los fenantrénicos, que se analizará en este capítulo son: la morfina y la codeína, que corresponden a hipnoanalgésicos naturales con acción sobre el músculo liso con acción analgésica diferenciada.
MORFINA
La morfina o tevaina (dimetilmorfina) es el principal alcaloide del opio descubierto por el boticario Sertürner en 1806, quien utilizó el nombre de morfina haciendo referencia a Morfeo (Dios de los sueños).2 Este fármaco es el analgésico más potente en el tratamiento del dolor de intensidad grave y moderada y su acción se facilita a través de la activación de receptores opioides específicos.3,9
Mecanismo de acción
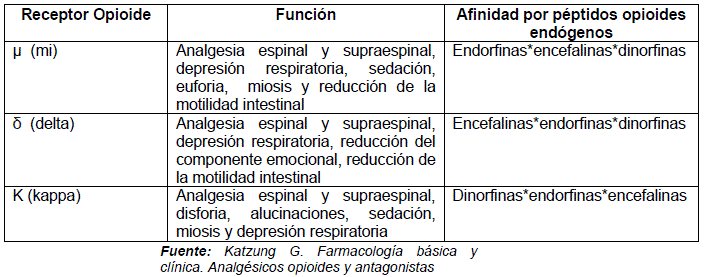
Para comprender la acción del opio, es necesario delimitar la labor de los receptores para opioides entre los que se encuentran:
Mu (μ) que se activa por la morfina y ocasiona analgesia supraespinal, depresión respiratoria, miosis, disminución de la motilidad intestinal, euforia, sedación, hipotermia y farmacodependencia.
Kappa (k), derivado de la ketaciclozina y media la analgesia espinal, sedación, miosis y depresión de reflejos motores
Omicron (o) que se activa por la SFK 10,047 o N-alilnormetazocina, receptor que no es tomado en cuenta porque guarda relación con alucinógenos como la ketamina y se ha vinculado al glutamato, pero es responsable de la presencia de discoria, alucinaciones, midriasis, taquicardia y activación respiratoria.
• Delta (δ), encargado de la analgesia supraepinal y espinal
La morfina presenta su acción analgésica en regiones donde el encéfalo presenta péptidos opiodes como la met-encefalina y leu-encefalina que interactúan con el receptor opioide,4 encontrándose además un cuarto receptor denominado delta (5) que hace que los péptidos muestren mayor afinidad que la morfina. De igual forma el hallazgo de (δ) -endorfina (1 y 2) con afinidad para el receptor μ y la dinorfina A con afinidad por el receptor K y de sus subtipos, han permitido una mejor comprensión de la acción de la morfina.
Las acciones directas se relacionan al cierre del canal del calcio en la región pre sináptica de las neuronas primarias encargadas de la conducción de señales nociceptivas, disminuyendo la liberación de neurotransmisores, y activación de canales de potasio en la neurona postsináptica de las vías conductoras del dolor, con la consiguiente hiperpolarización, que aparentemente bloquea la transmisión del dolor.5 La morfina actúa a nivel del sistema nervioso central (SNC), modificando la percepción del dolor; imitando a los pépticos opioides endógenos (endorfinas, encefalinas y dinorfinas), mediante la interacción con receptores específicos opiodes.6,11
Farmacocinética
La morfina se administra por diferentes vías, absorbiéndose a los 15 minutos por vía IM y SC, mientras que por vía oral la absorción es en un tiempo aproximado de 1 h., distribuyéndose por los tejidos a los 5 min., de ser administrada. Su difusión es de 1-6,2 l/kg, alcanzando concentraciones máximas en bilis y orina a las 3 h, llegando a una vida media de 1,7-4,5. Una vez ingresado al torrente circulatorio se une a proteínas en un 12 a 34% requiriéndose un nivel óptimo proteico en sangre, ya que en casos de hipoalbuminemia, su fijación estará considerablemente disminuida. Se metaboliza es en el hígado a partir del ácido glucorónico y su excreción se realiza por orina en un 12%, bilis 7%, pudiendo existir recirculación enterohepática que permite la reabsorción de la droga.7
El tiempo de vida, hace que su uso repetitivo durante el día en casos de pacientes con dolor crónico lleve a la dependencia, por lo que se recomienda el uso oral, aunque su biodisponibilidad baja se constituye en una limitante terapéutica, mientras que la vía parenteral requerirá de dosis mayores, para el efecto deseado, con todos los efectos colaterales consiguientes.
Algunos estudios han permitido describir el uso sublingual de esta droga, disminuyendo así el paso de metabolización hepática, disminuyendo de este modo la dosis y el tiempo de administración, así como los efectos indeseables. En estos casos se menciona la administración conjunta de antidepresivos que mejoran el efecto analgésico de la morfina.
Acciones Farmacológicas
La acción más importante de la morfina es la sedación del dolor moderado a severo de carácter agudo o crónico, acción producida por bloqueo de la sensación dolorosa a nivel del sistema nervioso central (SNC). De igual forma su acción sobre el aparato digestivo es de suma importancia, provocando aumento del tono y disminución de la motilidad intestinal.8 Sin embargo ésta droga no solo produce acciones en los sistemas antes mencionados, encontrándose también efectos sobre el sistema neuroendócrino estimulando la secreción de la hormona adrenocorticotropa (ACTH), somatotrópica, prolactina y la hormona antidiurética e inhibiendo la secreción de la hormona folículo estimulante (FSH) y luteinizante (LH).
A nivel del aparato respiratorio la morfina provoca depresión respiratoria por su acción sobre los receptores μ y δ situados en las neuronas de los núcleos bulbo protuberancial.
Otros efectos resultantes del uso de la droga son: la sedación o euforia, liberación de histaminas endógenas, estimulación anómala de los centros reguladores del movimiento a nivel central, etc.
Efectos colaterales
Los efectos colaterales, corresponden principalmente a la farmacodendencia cuando su uso es prolongado, además de inhibición del centro respiratorio provocando en los recién nacidos parálisis respiratoria y en los adultos condiciona la presencia de enfisema pulmonar, recomendándose tener cuidado en casos de asma o enfermedad pulmonar obstructiva crónica (EPOC). De igual forma la depresión del centro oculomotor a nivel central provocará miosis, que en casos de intoxicación o coma medicamentoso se convierte en dilatación pupilar marcada.
En el aparato digestivo, el estreñimiento se produce como resultante de bloqueo de la motilidad, asociándose a disminución de secreciones biliares y pancreáticas.9 La reducción del metabolismo hepático en ancianos, puede llevar a intoxicación por morfina, debiéndose ajustar la dosis.
Por su parte en el aparto cardiovascular presentará vasodilatación por liberación de histamina, depresión del centro vasomotor y la disminución contráctil de los músculos de los vasos sanguíneos.
Se recomienda igualmente tomar precauciones en mujeres embarazadas, niños y personas que tengan insuficiencia hepática y renal.
Vías de Administración y Dosis

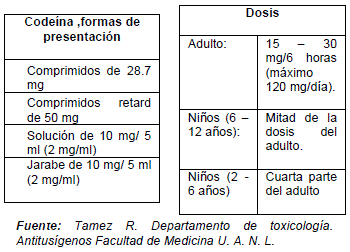
CODEINA
La codeína también denominada metilmorfina es un alcaloide natural que se encuentra en el opio, denominada así por su origen (cabeza de adormidera). Fue descubierta en 1832 por Pierre Robiquet, químico francés como una prodroga que se convierte metabólicamente en morfina.10 Este medicamento tiene poca afinidad por los receptores opioides por lo que su potencia y eficacia analgésica son inferiores al de la morfina, razón por la cual se indica en casos de dolor leve y moderado, requiriendo la mayor parte de las veces la asociación con otros medicamentos (ASA, acetaminofen) para potenciar su acción.11,14
El efecto principal de la codeína es la sedación de la tos, teniendo también acción sedante del SNC de baja intensidad, así como la disminución de la motilidad intestinal.12
Farmacocinética
Este medicamento se administra por vía oral, absorbiéndose rápidamente por el tracto gastrointestinal, luego de haberse fijado levemente a las proteínas plasmáticas (10%) alcanza su concentración máxima al cabo de 1 h. Su vida media es de 3-5 horas metabolizándose en el hígado a través de reacciones de O-desmetilación, N-desmetilación y conjugación con glucurónido , dando origen a la morfina y norcodeína que se excretan por la orina.13,10 Puede haber excreción por leche materna lo que limita su uso en la lactancia.
Efectos colaterales
La codeína es un irritante gástrico por lo que su uso en pacientes con antecedentes de ulcera gástrica o hemorragia digestiva están limitados. Del mismo modo al ser sedante de la tos, no se recomienda su uso en pacientes portadores de asma bronquial o rinitis alérgica. Este medicamento como ya se mencionó anteriormente, provoca sueño leve, recomendándose no manejar vehículos durante su uso.14 Al igual que la morfina su uso prolongado puede llevara farmacodependencia.
Formas de presentación y Dosis
NOTAS
1 Univ. Tercer Año Facultad de Odontología UMSA
2 Univ. Tercer Año Facultad de Odontología UMSA
BIBLIOGRAFIA
1. Goodman A. Las Bases Farmacológicas de la Terapéutica. Analgésicos Opioides. Cap 23. 10ma edición. México. Editorial McGraw-Hil Interamericana. 2003:577 - 596. [ Links ]
2. Patiño N. Farmacología Médica. Analgésicos Opioides. Cap. 22. 1ra edición. México. Editorial Médica Panamericana. 2008: 280 - 283. [ Links ]
3. Flores J., Armijo J., Farmacología Humana. Fármacos analgésicos opioides. Cap. 25. 4ta edición. Barcelona-España. Editorial Elsevier. 2003:461 -468. [ Links ]
4. Katzung G. Farmacología básica y clínica. Analgésicos opioides y antagonistas.. Cap. 31 10a edición. México. Editorial El Manual Moderno S. A. 2007:503-516. [ Links ]
5. TrIpathi K.D.; Farmacología en Odontología Fundamentos. Analgésicos y antagonistas opioides. Cap. 24 1ra edición. Madrid-España. Editorial Médica Panamericana. 2008: 352-361. [ Links ]
6. Page C. Curtis P. Walker M. Farmacología Integral. Fármacos en Odontología. Cap. 20. 3ra edición. Madrid-España. Editorial Harcourt Brace de España S. A. 1998: 383 -397. [ Links ]
7. Velásquez.; Lorenzo P.; Moreno A.; Farmacología Básica y Clínica. Fármacos analgésicos opioides. Cap. 12. 18va edición. España. Editorial Médica Panamericana. 2008: 213 - 222. [ Links ]
8. Rang H.P.; Dale M.M.; Ritter J.M.; Moore P. K.; Farmacología. Analgésicos. Cap. 40. 5ta edición. Madrid España. Editorial Elsevier S.A. 2004:571 -579. [ Links ]
9. Page C. Curtis P. Walker M. Farmacología Integral. Fármacos y sistema nervioso central. Cap. 7. 3ra edición. Madrid-España. Editorial Harcourt Brace de España S. A. 1998: 148-149. [ Links ]
10. Alfonso R. Farmacia. Drogas Analgésicas, Antipiréticas y antiinflamatorias. Cap. 83. 20a edición España. Editorial Medica Panamericana S. A. 2003: 1717 - 1720. [ Links ]
11. Pérez H. Farmacología y Terapéutica Odontológica. Analgésicos opiáceos. 2da edición. Editorial Médica Celsus. 2005: 189 -198. [ Links ]
12. Lullman H. Farmacología texto y atlas. Fármacos antinociceptivos. 6ta edición. Madrid-España. Editorial Médica Panamericana. 2010: 194 -198. [ Links ]
13. Litter M. Compendio de Farmacología. Hipnoanalgésicos naturales y sintéticos. Antagonistas de los opiáceos. Cap. 13 9na edición. Editorial El Ateneo. 2010: 158 - 167. [ Links ]
14. Ciancio S. Farmacología Clínica para Odontólogos. Analgésicos. Cap. 5. 3ra edición. España Editorial Manuel Moderno. S.A. 1990: 108-114. [ Links ]
15. Physician's D. Farmacología. Montvale, NJ: Medical Economics. 49a edición. 1995. Fecha de acceso: 02 de octubre del 2012; disponible en: http://www.uam.es/departamentos/medicina/anesnet/agenda/farmacologia/ morfina.htm [ Links ]
16.Tamez R. Antitusígenos. Departamento de toxicología. Facultad de Medicina U. A. N. L. Fecha de acceso: 02 de octubre del 2012; disponible en : https://docs.google.com/viewer?a=v&q=cache:ZJmw0xMDvQoJ:etableros.com/farmacologia/farma/diapos/files/antitusigenos.ppt+antitusigenos+departamento+de+toxicologia [ Links ]^rND^sEscobar Aguilar^nAlex Hugo^rND^sEsprella Rojas^nJavier Alejandro^rND^sEscobar Aguilar^nAlex Hugo^rND^sEsprella Rojas^nJavier Alejandro^rND^sEscobar Aguilar^nAlex Hugo^rND^sEsprella Rojas^nJavier Alejandro
ARTICULO
PENICILINAS
Escobar Aguilar Alex Hugo 1
Esprella Rojas Javier Alejandro 2
RESUMEN
En cualquier tipo de infección, la presencia de un gran número de especies bacterianas, son las responsables de producir distintas patologías que desencadenan efectos desfavorables y en ocasiones mortales para el organismo vivo. Es por eso que se crean los antibióticos, los cuales cumplen la función principal de suprimir y evitar la proliferación de las colonias microbianas.
Los antibióticos constituyen una familia numerosa e importante dentro de la farmacología. Debido a la aparición de nuevas patologías y microorganismos cada vez más resistentes, es que año tras año se realizan innumerables investigaciones para modificar los antibióticos y conseguir mejores propiedades terapéuticas. Un grupo muy destacado de la familia de los antibióticos, son las penicilinas, que cuentan con varias propiedades farmacológicas, entre una de ellas, inhibir el desarrollo bacteriano.
Las penicilinas son uno de los antibióticos más utilizados hoy en día, debido a que presenta una baja toxicidad para el organismo, aunque ciertas personas pueden desarrollar procesos alérgicos, pese a ello, son fármacos de primera elección ante las enfermedades infecciosas.
PALABRAS CLAVES
Antibiótico.Betalactámico. Penicilina
INTRODUCCION
Las penicilinas fueron descubiertas en 1928, por Alexander Fleming, mediante el aislado y el cultivo puro de un hongo del genero Penicillium, demostrando la producción de una sustancia bacteriana a la que denominó Penicilina, que no era tóxica para los animales de laboratorio. Florey y Chain, posteriormente, al determinar su uso en seres humanos, elaboraron un método para producirlo en gran escala.1-2
Actualmente las penicilinas constituyen uno de los antibióticos de mayor eficacia en infecciones, debido a su acción sobre la pared celular bacteriana, inhibiendo la síntesis de la misma.1-2
MECANISMO DE ACCION
Las penicilinas como cualquier antibiótico betalactámico, intervienen sobre la entidad bacteriana, inhibiendo la síntesis del péptidoglucano localizado en la pared celular del microorganismo.2-3
El objetivo de la penicilina es llegar a las proteínas fijadoras de penicilina (PFP), distribuidas en la membrana bacteriana, entre estas proteínas se mencionan la transpeptidasa, carboxipeptidasa y endopeptidasa, cuya función es catalizar la reacción de transpeptidación.2 La finalidad de este proceso es formar un enlace cruzado entre dos moléculas de péptidoglucano el cual le da la rigidez a la pared bacteriana. La penicilina al unirse con las proteínas fijadoras de penicilina, inhibe la reacción de transpeptidación, produciendo la muerte de la bacteria.3
ESTRUCTURA QUIMICA
Todas las penicilinas presentan en su estructura, un núcleo activo, el acido 6-aminopenicilánico, el cual está formado por un anillo betalactámico y un anillo tiazolidínico, formando una sola estructura y una cadena lateral, que no es definida.4
La función del anillo tiazolidínico es la de proteger al anillo betalactámico que le da a la penicilina su propiedad antibacteriana, siendo éste el lugar donde actúan las betalactamasas inutilizando a las penicilinas.4-5
La cadena lateral aparte de dar a las penicilinas varias de sus propiedades farmacológicas, permite la combinación con un inhibidor de betalactamasas, como el ácido clavulánico, el sulbactam, como inhibidores de betalactamasas.6-7
CLASIFICACION, DOSIS, VIAS DE ADMINISTRACION E INDICACIONES
Las penicilinas se pueden clasificar según el origen que tengan en tres importantes grupos: penicilinas naturales, biosintéticas y por último las semisintéticas.6
1. PENICILINAS NATURALES
Las penicilinas naturales son aquellas en las que no se realiza una preparación previa en un laboratorio y ninguna intervención tecnológica. Se obtienen por la fermentación, en un medio de cultivo, de un hongo conocido como Penicilium chrysogenum, dando como resultado a la Bencil penicilina G y sus derivados que son cuatro los cuales tienen indicaciones y dosis que las diferencian entre sí.6
2. PENICILINAS BIOSINTETICAS O ACIDORRESISTENTES
Debido a la posibilidad de dar un agregado a los medios de cultivo de la penicilina G, se pueden mejorar las propiedades y el rendimiento de la bencil penicilina; éste agregado lo constituye el ácido fenoxiacético que en la fermentación de los medios de cultivo del hongo Penicilium chrysogenum, da como resultado a la fenoximetil penicilina o penicilina V, la cual tiene la gran ventaja de ser insoluble en un medio ácido y soluble en un medio alcalino, lo que significa que no es destruida por los jugos gástricos.6
La fenoximetil penicilina V se administra por vía oral y está indicada en infecciones producidas por Streptococcus pyogenes. Su dosis en adultos es de 500 mg cada 6 horas por 7 días, mientras que la dosis pediátrica es de 125 a 250 mg Kg peso /día.6-7
3. PENICILINAS SEMISINTÉTICAS
Las penicilinas semisintéticas constituyen el grupo más numeroso puesto que su elaboración se realiza a partir del ácido 6-aminopenicilánico, que se obtiene también de la fermentación de los cultivos del hongo Penicilium chrysogenum. De acuerdo a su acción se dividen en dos grupos: las peniclinas resistentes a las beta lactamasas y las penicilinas mal llamadas de amplio espectro.8
Penicilinas resistentes a las beta lactamasas:
Este grupo presenta en su estructura una cadena lateral que le brinda protección ante la ruptura del anillo beta lactámico y evita la inactivación producida por la enzima beta lactamasa que presentan las bacterias, en especial estafilococos resistentes a las penicilinas naturales.6
Penicilinas mal llamadas de amplio espectro:
Son llamadas de ésta forma debido a que poseen un espectro mayor que las demás penicilinas, pero no superan a los antibióticos que realmente son considerados de amplio espectro.7
Como cualquier otra droga, la penicilina, sufre ciertas modificaciones en su concentración al momento de ser absorbidas por el organismo, esto con el fin de generar una respuesta.6-7
PENICILINAS NATURALES
PENICILINAS SEMISINTETICAS

PENICILINAS SINTETICAS
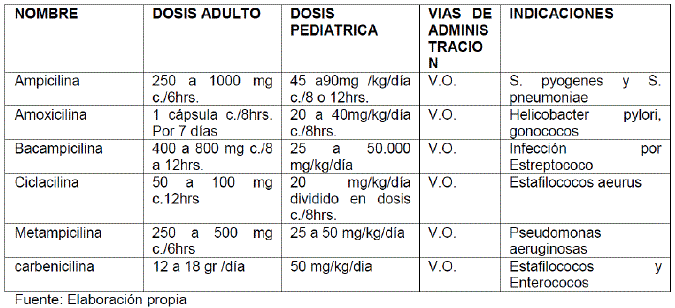
FARMACOCINETICA Y FARMACODINAMIA
Las penicilinas son administradas por diferentes vías y se absorben a nivel gastrointestinal. Sufren de la biotransformación en el hígado y su vía principal de excreción es la vía renal, pero también pueden ser eliminadas por medio de la bilis, la saliva y por la leche materna. Actúan sobre determinados grupos de microorganismos a lo que se denomina espectro antimicrobiano. Cuando las penicilinas actúan In Vitro, lo hacen fundamentalmente sobre los cocos Gram positivos y Gram negativos, sobre bacilos Gram positivos y muy poco sobre los Gram negativos. Dentro de los cocos Gram positivos se encuentran los Streptococcus beta hemolítico o pyogenes, a los Streptococcus alfa hemolítico o viridans y los Staphylococcusaeurussensibles.4-7
En el grupo de los cocos Gram negativos están presentes laNeisseria gonorrheaey a la Neisseria meningitidis. Sobre los bacilos Gram negativos las penicilinas actúan muy poco a excepción de la bacteria Melaninogenicum, la bacteria anerobia Actinomices israelii y el Treponema pallidum. Es de mucha importancia conocer que las penicilinas no actúan sobre el bacilo tuberculoso o de Koch.4-6
RESISTENCIA BACTERIANA
El mal uso de los antibióticos genera mecanismos de resistencia bacteriana resultantes del cambio cromosómico o de material genético bacteriano, a partir de tres mecanismos.2-3
1. Alteración de las proteínas blanco o fijadoras de la penicilina (PFP):
Las proteínas localizadas en la membrana citoplasmática de los microorganismos se unen por medio de enlaces covalentes a los betalactámicos.2-3
2. Inactivación del antibiótico por enzimas de tipo betalactamasas producidas por ciertas clases de bacterias: Se han identificado más de 100 clases de betalactamasas; la síntesis de las mismas es mediada por factores genéticos cromosómicos, los cuales presentan una síntesis continua de la enzima o sólo en presencia de la penicilina.2-3
La producción de la enzima separa el anillo betalactámico y genera derivados, uno de los más comunes y el de mayor importancia clínica entre las bacterias patógenas, es el ácido peniciloico.2-3
3. Disminución de la permeabilidad de la pared bacteriana por reducción de las porinas: Esta disminución de la permeabilidad esté relacionada con bacterias Gram negativas más que con las Gram positivas; debido a su resistencia, ya que poseen en su estructura porinas (proteínas específicas), las cuales confeccionan canales, por donde las moléculas hidrofílicas relativamente pequeñas de las penicilinas no obtienen el acceso a las PFP.2-3
CONTRAINDICACIONES
Las penicilinas como todo fármaco, puede producir reacciones de hipersensibilidad en el paciente, al detectarse que es alérgico a las penicilinas, se busca y selecciona otro fármaco.9-10
La insuficiencia renal es una patología en la cual está contraindicado el uso de la penicilina, debido a que las vías renales son el medio de excreción.10
EFECTOS ADVERSOS
Las penicilinas en general tienen poca probabilidad de producir efectos tóxicos directos, a diferencia del efecto proconvulsionante que ocurre cuando se administra por vía intratecal además de la hepatotoxicidad, dosis dependiente.9
La más común es la reacción de hipersensibilidad generada por la degradación de las penicilinas, cuyos productos entran en combinación con proteínas del huésped produciendo una reacción antígeno - anticuerpo. Es frecuente observar erupciones a nivel cutáneo, acompañado en la mayoría de los casos de fiebre. Un resultado poco probable pero muy peligroso es la evolución a un shock anafiláctico, que en la mayoría de los casos resulta mortal.9-10
NOTAS
1 Univ. Tercer Año Facultad de Odontología UMSA
2 Univ. Tercer Año Facultad de Odontología UMSA
BIBLIOGRAFIA
1. Pérez. H. Farmacología y Terapéutica Odontológica. 2a edición. Editorial Amolca. Argentina.; 2005; 301,302,303, 304. [ Links ]
2. Tripathi. K.D. Farmacología en odontología.1a edición. Editorial Médica Panamericana. Buenos Aires - Argentina; 2008; 404, 405, 407. [ Links ]
3. Rang. H.P. Dale. M.M. Ritter J.M. Moore. P. K. Farmacología. 5a edición. Elsevier. Madrid - España; 2004; 640. [ Links ]
4. Vallory. S.J. Vademecum de Odontología. 1a edición. Vademecum de Bolsillo S.R.L. Buenos Aires -Argentina; 1997; 362, 363. [ Links ]
5. Mascaro.J.M. Diccionario Medico. 3a edición. Salvat Ciencia y Cultura Latinoamericana. México; 2000; 507. [ Links ]
6. Laurence. L. Goodman y Gilman. Las Bases Farmacológica de la Terapéutica. 11a edición. MG Grill. España; 2006; 256, 257, 258. [ Links ]
7. Gennaro. A.R. Farmacia: Remington. 20a edición. Médica Panamericana. Buenos Aires - Argentina. 2003; 215, 216,214 [ Links ]
8. Rang. H. P. Farmacología. 5a edición. Harcourt/Elsevier, Madrid - España. 2000; 485, 486. [ Links ]
9. Smith. C. M. Farmacología 1a edición. Médica Panamericana. Buenos Aires -Argentina. 1998; 129. 130 [ Links ]
10. Martin. E. Farmacia practica de Remington. 2a edición. UTEHA. España 1965; 255 [ Links ]^rND^sEnríquez Torrez^nJimmy Steve^rND^sDuran Zúñiga^nRocío Lizeth^rND^sEnríquez Torrez^nJimmy Steve^rND^sDuran Zúñiga^nRocío Lizeth^rND^sEnríquez Torrez^nJimmy Steve^rND^sDuran Zúñiga^nRocío Lizeth
ARTICULO
MACROLIDOS Y AMINOGLUCOSIDOS
EnríquezTorrez Jimmy Steve 1
Duran Zúñiga Rocío Lizeth 2
RESUMEN
Los macrólidos son antibióticos naturales, semisintéticos o sintéticos obtenidos a partir de productos metabólicos del Streptomyces spp en 1942 , cuya acción fundamental es la inhibición de la síntesis proteica bacteriana. Durante los últimos años, la modificación del número de átomos de su cadena ha permitido la aparición de nuevos medicamentos que permiten menores efectos colaterales y mejor cobertura antibiótica.
Por su parte, el grupo de los aminoglucósidos, incluidos desde 1943 a partir de un producto metabólico del Strptomyces griseus, corresponden a medicamentos bactericidas de espectro reducido, y al igual que el grupo antes descrito inhiben la síntesis proteica de las bacterias atacando principalmente a bacilos aerobios Gram negativos. En la actualidad se ha disminuido su uso debido a la nefrotoxicidad y ototoxicidad, que son reversibles en sus fases tempranas y con la interrupción de la administración del medicamento. Sin embargo en algunos casos continúan siendo antibióticos de elección, acompañados de otros fármacos como la penicilina, vancomicina y cefalosporina, que sirven para potenciar su acción.
PALABRAS CLAVES
Macrólidos. Aminoglucósidos. Antibióticos. Gentamicina. Bacterias Gram negativas.
INTRODUCCION
Los macrólidos son una extensa familia de antibióticos logrados a partir de productos metabólicos del Streptomyces spp., que fueron descubiertos en 1942 por Gardner y Chain, quienes obtuvieron el primer compuesto del grupo, la Pricomicina. Una década después en 1952 en los laboratorios Eli Lilly, McGuire y colaboradores, obtienen la iloticina a partir de una cepa de Streptomyces eruthraeus, obtenida de un ejemplar de suelo recogido en el archipiélago Filipino a través del cual se obtuvo el primer macrólido denominado eritromicina.
Por su parte los aminoglucósidos fueron introducidos en 1943 luego de que Waksman aisló de la cepa de Streptomyces griseus a la estreptomicina, que le valió su nominación al nobel de medicina, después de tan histórico descubrimiento se aislaron nuevos medicamentos con actividad antibacteriana mejorada, y menores efectos colaterales.
MACROLIDOS
Los macrólidos son una variedad de antibióticos de espectro reducido, formados por un anillo de lactona macrocíclica. La diversidad de compuestos, que integran ésta familia se basan en la cantidad de átomos de su molécula y cada conjunto tiene características químicas y biológicas diferenciales, propias sobre su solubilidad en el agua o su resistencia en medios ácidos.1,4,7,12.
CLASIFICACION
Estos medicamentos químicamente caracterizados por su anillo macrocíclico lactónico, se catalogan de acuerdo al número de azúcares que varían su átomo, de ésta manera la sustitución de cladinosa que ocupa la posición 3 por un grupo cetónico ha dado lugar a una nueva familia denominada cetólidos. Si el representante del grupo de 14 átomos macroclícos es la eritromicina, el prototipo de los cetólidos es la telitromicina.2,4,7,8,12
Los grupos así originados, se basan en la cantidad de átomos que tienen, de ésta forma se pueden mencionar a:1,14
MECANISMO DE ACCION
Estos medicamentos tienen acciones bactericidas y bacteriostáticas :2,3,6,7,10La acción bactericida, dependerá de la fase de crecimiento del germen inóculado y el pH del medio donde llegará a actuar. El mecanismo de acción bacteriostática que ofrecen los macrólidos se relaciona a la detención de las síntesis proteica ribosómica de las bacterias al unirse por puentes de hidrógeno, en forma reversible al dominio V del ARN de la unidad 23S y a algunas unidades ARNr 50 S, impidiendo la reacción de translocación en la cadena peptídica que crece, desplazándose del sitio de recepción al del donador, razón por la cual se prohíbe su uso con drogas que compitan por dicho sitio ribosomal (clindamicina, cloranfenicol), es decir bloqueando la salida proteínica del ribosoma.4,8
FARMACOCINETICA
Con excepción de la Azitromicina, todos los macrólidos se metabolizan en el hígado, utilizando la vía del sistema enzimático citocromo P-450, su pico sérico depende de la dosis y el uso de otros agentes de metabolización hepática, que pueden llevar a su saturación.4Laincorporación celular se realiza por transporte activo dependiente dela concentración del calcio, logrando en el citoplasma celular niveles mayores a las plasmáticas (de hasta 7 veces mayor en la azitromicina y telitromicina), almacenándose la mayor parte del medicamento en los fagolisosomas de las células. Su difusión a través de las meninges es pobre, al igual que la placenta y feto, llegando a la saliva, leche materna y vías respiratorias en concentraciones mayores al 50% de las halladas en suero.4
La eliminación será realizada por vía biliar como metabolitos activos, con concentraciones mayores a la sérica, mientras que la eliminación por víaurinaria es inferior al 10% (aexcepción de la claritromicina que llega hasta el 30%).4
AMINOGLUCOSIDOS
Cualitativamente las propiedades de los aminoglucósidos son similares entre sí, son antimicrobianos que actúan básicamente a nivel ribosomal y atacan a bacterias metabólicamente activas inhibiendo su posterior desarrollo.A pesar de sus efectos tóxicos sobre el oído y el riñón, son aún muy usados por su rápido efecto bactericida, baja resistencia, dosis dependiente y bajo costo.
ESPECTRO DE ACCIÓN
El espectro de acción se encuentra resumido en la siguiente tabla:

DOSIS, VÍAS DE ADMINISTRACIÓN Y EFECTOS COLATERALES12
CLASIFICACION Y ESTRUCTURA QUIMICA
La estructura química de los aminoglucósidos se compone de aminoazúcares que se encuentran unidos por enlaces glucosídicos a un alcohol especifico cíclico hexagonal con grupos amino. De acuerdo al componente aminociclitol se clasifican de la siguiente manera:

MECANISMO DE ACCION
El mecanismo de acción de los aminoglucósidos se basa, en la interacción con la superficie externa de la membrana celular bacteriana, luego el transporte por la membrana interna y por último la unión a la subunidad 30S de los ribosomas, inhibiendo la síntesis proteica. Los aminoglucósidos son bactericidas rápidos y son potenciados por los fármacos que interfieren en la síntesis de la pared celular7. Sin embargo el mecanismo preciso del cual depende el efecto letal sobre las bacterias no está completamente descrito \ La principal utilidad de la gentamicina está en su eficacia contra bacilos Gram negativos aerobios (Pseudomona aeruginosa, Echericha coli. Klebsiella y especies de Preoteus).
FARMACOCINETICA
Estos medicamentos son de administración parenteral, tienen una absorción completa por vía intramuscular, alcanzando su máxima concentración aproximadamente a los 30 a 90 minutos, por vía intravenosa se alcanza entre 60 y 90 minutos. Son excretados por filtración glomerular.
La dosis y vía de administración se resumen en la tabla:

NOTAS
1 Univ. Tercer Año Facultad de Odontología UMSA
2 Univ. Tercer Año Facultad de Odontología UMSA
BIBLIOGRAFIA
1. Guía ADA / PDR de Terapéutica Dental / Asociación Dental Americana & Thomson PDR - Madrid, Editorial Medica Ripano 4ta edición. 2008; 189 -193.
2. Velásquez; De Lorenzo.; A. Moreno; Lizasoain P.A. Farmacología básica, 17ma edición. México.Editorial Panamericana septiembre 2004 reimpresión mayo 2005: 809 - 817. [ Links ]
3. Sanz de Miguel MP; Sancho Gracia E. Reacciones Adversas Psiquiátricas asociadas a nuevos macrolidos. A propósito de tres casos; Notas clínicas, Revista pediátrica de atención: 249-253 [ Links ]
4. Tripathi Farmacología fundamentos, 1ra Edición. Madrid España .Editorial Médica Panamericana 2008: 435 -441 [ Links ]
5. Netter R. Robert B, M.Rawls S, Farmacología Ilustrada, 1ra edición española, Barcelona España. Editorial Elsevier. 2008: 315-316 [ Links ]
6. Pérez Torrez H, Farmacología y Terapéutica Odontológica. 2da edición. Editorial Amolca. 2005: 287 -321. [ Links ]
7. Rodríguez Carranza R; Vademécum 3ra edición. Editorial MC Graw- Hill Interamericana. México .1999: 49 -50, 185 - 187,330 - 331, 343 - 344, 426, 429, 525 - 527, 666 - 667, 832. [ Links ]
8. RaneH.P; DaleM.M, RitterJ.M; MooreP.K, Farmacología, 5ta edición, Madrid - España. Editorial Elsevier 2004: 635- 647 [ Links ]
9. Rodríguez M. Aminoglucósidos. EnfInfec y Micro. 2002;ene/mar 20 -30; 22 (1): 20-30. URL disponible en: www.medigraphic.com/pdfs/micro/ei-2002/ei021d.pdf. Fecha de acceso: 21 de septiembre del 2012. [ Links ]
10. Caballero J. Macrolidos. Rev Pac de Med Fam. 2007: 4(6): 149-153 URL disponible en: www.mflapaz.com/Revista_6/revista_6_pdf/11%20macrolidos.pdf. Fecha de acceso: 21 de septiembre del 2012. [ Links ]
11. Ciancio S.G. Priscilla C. Bourgault, Farmacología Clínica para Odontólogos, 3ra Edición, México DF. Editorial Manual Moderno S.A. de C.V., Bogotá 1990:57- 75. [ Links ]
12.Gundián Gonzales J., Barreto Penie, J., Ange M.; Rodríguez R., Pablo Pino Alfonzo P y Lim Alonso N. Acta médica 1998: (8)1: 71-74. bvs.sld.cu/revistas/act/vol8_1_98/act10198.htm [ Links ]
13. Molina J, Cordero E, Palomino J y Pacho'n J. Aminoglucósidos y polimixinas. URL disponible en: EnfermInfecc Microbiol Clin.2009; sep 21; 27(3):178-188 [ Links ]
14. Lucas M F, Mestorino M, Errecalde JO .Macrólidos: Novedades de unDclásico grupo de antimicrobianos. Analecta veterinaria. 2007; 27 (1): 36-45. URL disponible en: www.old.fcv.unlp.edu.ar/analecta/volumenes/contenido/macrolidos.pdf Fecha de acceso 21 sept.2012 [ Links ]^rND^sSanz de Miguel^nMP^rND^sSancho Gracia^nE^rND^sCaballero^nJ^rND^sGundián Gonzales^nJ^rND^sBarreto Penie^nJ^rND^sAnge^nM^rND^sRodríguez^nR^rND^nPablo^sPino Alfonzo P^rND^sLim^nAlonso N^rND^nLucas^sM F^rND^sMestorino^nM^rND^sErrecalde^nJO^rND^sGutierrez Quispe^nNeiza Laura^rND^sGutierrez Quispe^nNeiza Laura^rND^sGutierrez Quispe^nNeiza Laura
ARTICULO
CEFALOSPORINAS
Gutierrez Quispe Neiza Laura 1
RESUMEN
Las cefalosporinas son antibióticos de espectro reducido que presentan una estructura química similar a la de la penicilina, ya que ambas pertenecen a la familia de los betalactámicos y tienen la capacidad de actuar como quimioterápicos-bactericidas por medio de la inhibición de la síntesis del peptidoglucano, uno de los principales componentes de la pared bacteriana.
A estos fármacos se les atribuye la capacidad de atacar al microorganismo sin producir efectos tóxicos de importancia, sin embargo se menciona que su uso puede producir cierto grado de alteraciones cutáneas, renales o digestivas.
Estos antibióticos se clasificarán de acuerdo a su origen en naturales y semisintéticos, dentro del segundo grupo encontramos a las cefalosporinas de primera, segunda, tercera y cuarta generación.
Las diferentes presentaciones de estos fármacos, nos permiten usar dos tipos de administración: vía oral y parenteral, recomendándose ésta ultima por producir un efecto más rápido y una concentración sanguínea máxima que es alcanzada en pocos minutos.
Estos fármacos son considerados como tratamiento de segunda elección en casos en los que se presente resistencia bacteriana, y serán utilizados como antibióticos de acción terapéutica y profiláctica.
PALABRAS CLAVE
Cefalosporinas. Antibióticos. Quimioterápicos
INTRODUCCION
Las cefalosporinas son antibióticos que forman parte del grupo de los ß-lactámicos, que poseen un anillo ß-lactámico ligado a un anillo dihidrotiazínico, cuyo resultado es la constitución del núcleo cefem del que derivan todas las cefalosporinas.8
Existen diversas formas de clasificar a estos antibióticos, pero la más utilizada es aquella que agrupa a estos compuestos tomando en cuenta ciertas características como las propiedades estructurales, microbiológicas y desarrollo histórico; dándonos la clasificación de cefalosporinas de primera, segunda, tercera y cuarta generación.9
ANTECEDENTES HISTORICOS
En 1948, Brotzu aisló por primera vez al Cephalosporium acremonium, un hongo empleado como fuente inicial para la producción de cefalosporinas, que se obtuvo del agua de mar cercano a un sitio de descarga de aguas negras en la costa de Cerdeña. Los estudios microbiológicos que se realizaron con el cultivo de dicho hongo, mostraron su capacidad de inhibir la proliferacion in vitro del Staphilococcus aureus. Es así como se originaron las cefalosporinas naturales: C, Py N.1
Las cefalosporinas actualmente usadas son las de tipo semisintético, que se originan gracias a la modificación de las cadenas laterales de la cefalosporina C, dando como resultado el anillo 7-amino-cefalosporánico.2,4,9
CLASIFICACION
Desde un punto de vista clínico, para agrupar a estos fármacos se emplea la clasificación por generaciones, que divide a estos antibióticos según el desarrollo histórico y algunas características en su estructura y microbiología, en cefalosporinas de:
a) Primera generación. A este grupo pertenecen los siguientes antibióticos: Cefalexina, Cefradina, Cefadroxil, Cefazolina, Cefalotina, Cefapirina, Cefradina y Cefaloridina.
b) Segunda generación. Forman parte de esta generación los fármacos comoel:Cefaclor, Cefuroxima Axetil, Cefazoril, Cefatrizina, Loracarbef, Cefprozil, Cefamandol, Cefuroxima, Cefonicida, Ceforanida y el grupo de las Cefamecinas, éstas son: Cefoxitina, Cefotetan y Cefmetazol.
c) Tercera generación. En ésta clasificación se encuentra a: Cefixima, Cefprozil, Cefpodoxima, Ceftibuten, Cefdimir, Cefetamed-pivoxil, Cefotaxima, Ceftriaxona, Ceftizoxima, Moxalactzm, Cefpiramida, Cefzulodin; elsubgrupo de las Antipseudomonas, como: Ceftazidina y Cefoperazona; y los inmunoestimulantes, cuyo representante es la Cefodizima.
d) Cuarta generación. Estos antibióticos son: Cefpirome, Cefepime, Cefaclidina, Cefquinona, Cefzulodin.8
FARMACODINAMIA
Las cefalosporinas de primera, segunda y tercera generación tienen una farmacodinamia semejante, ya que actúan sobre microorganismos como los cocos y bacilos, que pueden ser de tipo Gram positivos, Gram negativos y espiroquetas.3
Los antibióticos que pertenecen a la segunda generación también actúan sobre las enterobacterias y a la farmacodinamia de las cefalosporinas de tercera generación se suma la acción sobre las pseudomonas y bacteroides.
Los fármacos que pertenecen al grupo de cefalosporinas de cuarta generación actúan ante enterobacterias resistentes, bacterias Gram negativas trofoespecíficas y pseudomonas aeruginosa.3
FARMACOCINETICA
Las cefalosporinas se administran casi exclusivamente por vía parenteral e intramuscular, pudiendo también administrarse por vía oral a excepción de las cefalosporinas de cuarta generación, cuya única vía de administración es la vía parenteral intravenosa.3
En general, éstos fármacos, tienen una concentración máxima por vía parenteral después de la inyección a los 30 a 75 minutos (en cefalosporinas de primera generación) y a los 30 minutos (en cefalosporinas de segunda y tercera generación).3
Los antibióticos que pertenecen a la cuarta generación tienen una vida media sérica registrada de dos horas.1
Todas las cefalosporinas son eliminadas por vía renal. La ceftriaxona y la cefoperazona se eliminan también por vía biliar. Son metabolizadas mediante acetilación la cefalotina y la cefotaxima.
INDICACIONES TERAPEUTICAS
Las cefalosporinas en general son eficaces como medicamentos terapéuticos y profilácticos, actuando sobre infecciones provocadas por los siguientes microorganismos: estreptococos, Estafilococos aureus, Klebsiella pneumoniae, Escherichia coli, Proteus mirabilis, Haemophilus influenzae, Moraxella catharralis.1,3 Una mejor manera de sintetizar las indicaciones terapeúticas y describirlas de una forma más específica, es a partir de la clasificación de las cefalosporinas por generaciones en:
a) Primera generación: son excelentes en infecciones de la piel y tejidos blandos, en infecciones respiratorias, urinarias y osteoarticulares. Además de su uso en dosis única antes de una intervención quirúrgica.1,11
b) Segunda generación: han quedado desplazadas por las de tercera generación, en el tratamiento de muchas infecciones, es por ésta razón que no deberán ser aplicadas en tratamientos empíricos de meningitis o neumonía. Sin embargo pueden utilizarse en tratamientos de infecciones respiratorias altas y bajas, administrando el medicamento por vía oral, en infecciones intraabdominales, enfermedad inflamatoria pélvica, e infecciones de pies diabéticos. Actuando sobre los siguientes microorganismos: Enterobacterias, Haemophilus influenzae, Moraxella catharralis.1, 11
c) Tercera generación: pueden estar combinadas con o sin aminoglúcósidos, son eficaces en el tratamiento de todas la formas de gonorrea y formas graves de la enfermedad de Lyme; además de combinadas con vancomicina y ampicilina son usadas en la meningitis, y pueden también ser usadas en casos de neumonía adquirida en la comunidad. Actúan sobre los siguientes microorganismos: Enterobacterias, Serratia marcescens, Pseudomona aeruginosa, Neisseria. gonorrheae, S. pyogens y S. aureus (cefotaxima). B. fragilis (ceftizoxima y cefoxitina).1
d) Cuarta generación están indicadas en el tratamiento empírico de infecciones nosocomiales, donde se anticipa resistencia a antibióticos.1
En el área de la odontología varios estudios han indicado que las cefalosporinas pueden ser utilizadas en el tratamiento de infecciones odontógenas causadas por el género Klebsiella, ya que se menciona que un 2% de las especies de microorganismos aislados en una caries dental pertenecen a este género de bacterias. También son utilizadas en casos donde se tratan infecciones producidas por estafilococos productores de penilicinasa o en pacientes que presentan reacciones alérgicas a la penicilina.
MECANISMO DE ACCION DE LAS CEFALOSPORINAS
Las cefalosporinas al igual que las penicilinas se unen por medio de enlaces covalentes al sitio activo de la enzima transpeptidasa, ambos medicamentos actúan como bactericidas en etapa de multiplicación bacteriana, sin embargo los autores mencionan que las cefalosporinas actúan sobre todo en la fase de crecimiento bacteriano.
Se debe resaltar que el principal mecanismo de acción de las cefalosporinas es la inhibición de la síntesis de la pared celular bacteriana y como consecuencia se logra inhibir la transpeptidasa, produciendo la lisis de la bacteria.2,3,6
REACCIONES ADVERSAS
Se observan reacciones de tipo inmediato como anafilaxia, broncoespasmo y urticaria. Con mayor frecuencia aparecen erupciones maculopapulares y después de varios días de administración se puede presentar también fiebre y eosinofilia.1
Ante la semejanza con la penicilina también puede producir reacciones alérgicas en caso de que el paciente sea alérgico a dicho fármaco, debido a la reacción cruzada que producen ambos medicamentos (cefalosporinas y penicilinas) al presentar una estructura similar, por ésta razón la administración se debe realizar con mucha cautela.1,3,6
En casos infrecuentes, las cefalosporinas han producido una depresión de médula roja, caracterizado por granulocitopenia.1
Después de administrar dosis de cefalosporinas mayores a 4g/día, se ha observado necrosis tubular renal y nefrotoxicidad.1,3
Pueden también producirse efectos como hemorragias, disfunción plaquetaria, intolerancia al alcohol, trombocitopenia y cuadros caracterizados por vómitos y diarrea en la administración oral de algunos de estos fármacos. Se atribuye a estos fármacos la reacción de nefrotoxicidad, ya que el riñón es el sitio de filtración y posterior eliminación.
CONTRAINDICACIONES
Estos fármacos están contraindicados en caso de alergia a las cefalosporinas y a la penicilina. Deben emplearse con sumo cuidado en pacientes que padezcan de alteraciones renales, tomando en cuenta que en algunos casos esto puede solucionarse disminuyendo la dosis de administración.3
NOTAS
1 Univ. Tercer Año Facultad de Odontología UMSA
BIBLIOGRAFIA
1. Hardman J., Limbird L. Las bases farmacológicas de la terapéutica. 10ma edición. México: Mc Graw Hill. 2002: 1207-1231. [ Links ]
2. Enid A, Donald C, Jhon A, Janes W. Farmacología y terapéutica odontológicas. 1ra edición. México: Nueva editorial Interamericana. 1985: 583-585. [ Links ]
3. Litter M. Compendio de farmacología. 3ra edición. España: El ateneo. 1984:679-718. [ Links ]
4. Rang H.P. Farmacología. 5ta edición. España: Elsevier. 2004: 620- 642. [ Links ]
5. Lasala A. Endodoncia. 3ra edición. España: Salvat Editores. 1979: 169-172. [ Links ]
6. Lullman H. Farmacologíatexto atlas. 6ta edición. España: Panamericana. 1990:252-255. [ Links ]
7. Greame S.A. Farmacología Clínica y Terapéutica. 2da edición. España: Salvat editores. 1980: 1040-1056. [ Links ]
8. Mella S., Zemelman C., Bello H., Domínguez M., González G., Zemelman R. Propiedades microbiológicas, clasificación y relación estructura-actividad de cefalosporinas e importancia de las cefalosporinas de cuarta generación. Rev Chil Infect ; 2001; 18 (1): 7-19. Fecha de acceso 30 de septiembre de 2012. URL disponible en: http://www.scielo.cl/pdf/rci/v18n1/art02.pdf [ Links ]
9. Zamora Marín R., Areu Regateiro A., Gundián J. y Colaboradores Cefalosporinas. Acta Médica 1998; 8(1): 40-7. Fecha de acceso 30 de septiembre de 2012. URL disponible en: http://bvs.sld.cu/revistas/act/vol8_1_98/act05198.pdf [ Links ]
10. Núñez Freile B.Las cefalosporinas. Fecha de acceso 30 de septiembre de 2012. URL disponible en: http://mvz.unipaz.edu.co/textos/lecturas/preclinica/cefalosporinas2.pdf [ Links ]^rND^sMella^nS^rND^sZemelman^nC^rND^sBello^nH^rND^sDomínguez^nM^rND^sGonzález^nG^rND^sZemelman^nR^rND^sZamora Marín^nR^rND^sAreu Regateiro^nA^rND^sGundián^nJ^rND^sHoyos Serrano^nMaddelainne^rND^sHoyos Serrano^nMaddelainne^rND^sHoyos Serrano^nMaddelainne
ARTICULO
QUINOLONAS. CARBAPENEMS
Hoyos Serrano Maddelainne1
RESUMEN
Las quinolonas y los carbapenems, son antibióticos bactericidas de amplio espectro, que han tenido un extenso desarrollo, debido a las cualidades farmacológicas de sus predecesores: el ácido nalidíxico y la tienamicina y a las necesidades terapéuticas de infecciones muy resistentes a antibióticos habituales.
Las quinolonas inhiben las enzimas (DNA-girasa y topoisomerasa IV) encargadas del recorte del DNA bacteriano para su posterior transcripción y replicación, impidiendola síntesis de proteínasnecesarias en la reparación, crecimiento y reproduccióny por consiguiente, produciendo muerte bacteriana. Por otro lado, los carbapenems a semejanza de otros antibióticos (3-lactámicos, se unen a proteínas ligadoras de penicilina dificultando la síntesis de la pared celular y produciendo lisis bacteriana.
El surgimiento y extensión de resistencia a las quinolonas las ha limitado en su uso en algunos casos, no obstante todavía se las puede utilizar en muchos procesos infecciosos cuyos agentes etiológicos son sensibles. Por otra parte la resistencia a los carbapenems es muy baja a comparación de las quinolonas, ya que su espectro antibacteriano es el más amplio disponible, de modo quepueden ser utilizados en infecciones en cualquier parte del organismo que hayan presentado dificultades a antimicrobianos habituales, convirtiéndose en los quimioterapéuticos de última elección.
PALABRAS CLAVE
Fluoroquinolonas. Carbapenems. Imipenem. Antibióticos de amplio espectro.
INTRODUCCION
El desarrollo del ácido nalidíxico y la tienamicina,han ampliado los horizontes de la quimioterapéutica, puesto que han permitido generar dos grandes familias de antibióticos: las quinolonas y los carbapenems, antibióticos de amplio espectro que se utilizan para tratar una diversidad de infecciones.
Las quinolonas se hallan disponibles desde hace cuatro décadas y debido a sus cualidades bactericidas, se las ha utilizado expansivamente, de manera que se han desarrolladoresistencias que las limitaen su querencia. Por otro lado la era de los carbapenems se halla en boga, ya que éstos son los antibióticos con el mayor espectro disponible, y en apenas 25 años se ha logrado mejorar las características de su primer miembro, el imipinem, con el desarrollo del ertapenem y doripenem, antimicrobianos con mayor vida media y menor metabolismo, lo que les otorga seguridad al momento de su administración.
QUINOLONAS
Las quinolonas integran una familia de antibióticos bactericidas de amplio espectro, que combaten contra bacterias Gram-positivas y Gram-negativas.1
ESTRUCTURA QUIMICA, CLASIFICACION Y ESPECTRO ANTIBACTERIANO
En 1962, los laboratorios de Sterling-Winthrop desarrollaron el primer compuesto de las quinolonas, el ácido nalidíxico, obtenido a partir de la cloroquina.2 En 1973 apareció la primera quinolona con un átomo de flúor: la flumequinolona, pero es en 1978 cuando se inicia la era de las quinolonas fluoradas con la síntesis del norfloxacino y otros numerosos compuestos.3 (cuadro N°1)
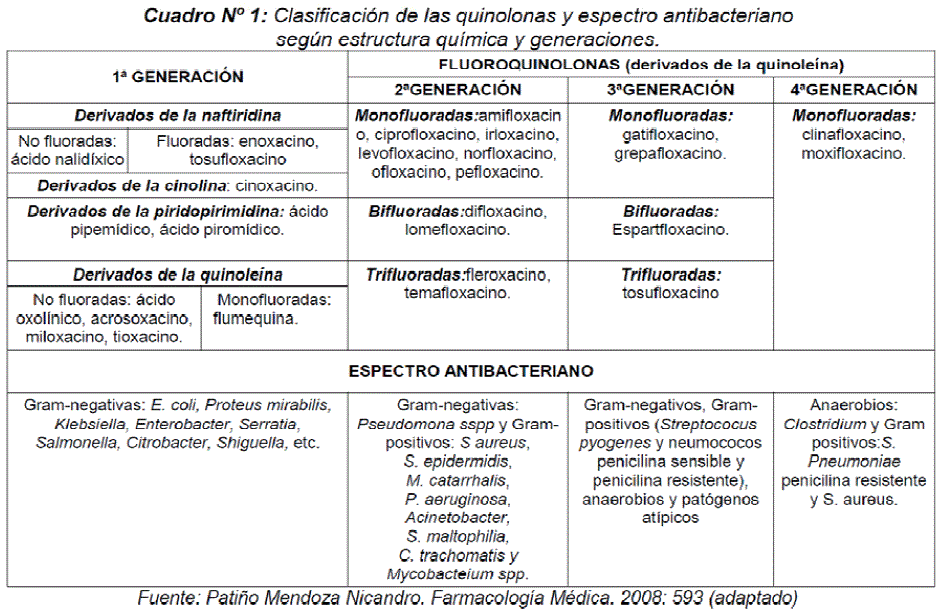
La estructura química está basada en el anillo 4-oxo-1, 4-dihidroquinoleína, del que derivan 4 grupos (naftiridina, cinolina, quinoleína y piridopirimidina) que se forman según las distintas sustitucionesde nitrógeno en los diferentes átomos: 1 y 8 para la naftiridina, 1 y 2 para las cinolinas, 1 para la quinoleína y 1,6 y 8 para la piridopirimidina. Cabe recalcar que la incorporación de un átomo de flúor en posición 6 y el grupo piperazínico heterocíclico en el 7, aumentan la actividad antibacteriana.3
Atendiendo a su estructura química, las quinolonas se las pueden clasificar en diversos grupos, pero también se las puede clasificar en generaciones, a continuación se detalla ambas clasificaciones integradas.3(cuadro N°1)
FARMACODINAMIAY FARMACOCINETICA
Esencialmente, se acepta que las quinolonas y fluoroquinolonas penetran a la bacteria a través del canal acuoso de las porinas, una vez dentro, en bacterias Gram-negativas (como la E. coli) inhiben la acción de la DNA-girasa, enzima encargada del recorte de la molécula del DNA para su posterior transcripción y replicación; de manera similar, en bacterias Gram-positivas (como S. aureus) las quinolonas bloquean la operación de la topoisomerasa IV, sustancia que separa las moléculas de DNA hijas entrelazadas, producto de replicación del DNA.1,2,4 La consecuencia del bloqueo de la actividad de éstas enzimas en la célula bacteriana, da lugar a la inhibición de la traducción para la síntesis de proteínas necesarias en la reparación, crecimiento y reproducción bacteriana, de este modo la inhabilitación prolongada de ambas enzimasconduce a la muerte de la célula bacteriana.1
Las quinolonas de primera generación y las fluoroquinolonas presentan una buena absorción tras su administración oral, las últimas, alcanzan su tiempo máximo al cabo de 1-3 horas.2,3,5 (cuadro N° 2) Estos fármacos se concentran en muchos tejidos, especialmente en riñón, próstata y pulmón, no así en SNC, a excepción del pefloxacino y oxofloxacino, que alcanzan en el LCR concentraciones del 40% y 90% respectivamente de sus cifras séricas.6
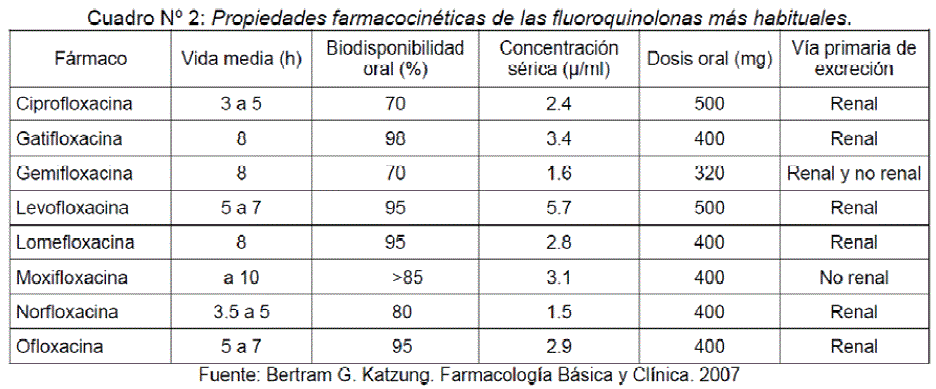
La mayoría de las quinolonas atraviesan la placenta y se concentran en el líquido amniótico, se eliminan por vía renal (ácido pipemídico, ofloxacina, levofloxacina), otras por vía fecal (moxifloxacina) y otras por ambas vías (norfloxacina, ciprofloxacina).(cuadro N° 2) La ciprofloxacina, oxofloxacina y pefloxacina han sido detectadas en la leche materna, luego de un uso rutinario del medicamento, por lo que deben evitarse en la lactancia, debido a que puede producir defectos en el desarrollo óseo y articular del recién nacido.1,3,6
RESISTENCIA BACTERIANA
La resistenciabacteriana es cruzada entre quinolonas de primera generación y entre fluoroquinolonas, pero habitualmente no existe resistencia cruzada entre ambos grupos, ni con otros antibióticos.2,3
La resistencia a las quinolonas puede surgir durante el tratamiento, especialmente en infecciones por Pseudomonas y Serratia, debido a mutaciones delos genes cromosómicos bacterianos que codifican para la DNA-girasa o topoisomerasa IV,lo cual impide la unión de la quinolona a estas enzimas.14En bacterias Gram-negativas se han descrito resistencias de bajo nivel por alteraciones en las porinas que hay en la membrana externa.17En plásmidos sólo se ha confirmado resistencia transferible en un número pequeño de cepas de Klebsiella pneuomniae, éste plásmido, lleva un gen que codifica un proteína denominada Qnr que de manera desconocida protege la DNA-girasa de la acción de las quinolonas.7Es importante destacar, que existe resistencia de los neumococos a estos antibióticos y que se adquiere de forma escalonada, produciéndose inicialmente una mutación en genes parC o gyrA que expresan sus enzimas diana topoisomerasa IV y DNA-girasa haciéndolos menos sensibles. Sin embargo, para que se produzca una resistencia completa, es necesaria una segunda mutación en el otro gen diana.8
INDICACIONES TERAPEUTICAS, REACCIONES ADVERSAS E INTERACCIONES
Según Rodríguez Consuelo9 las quinolonas y fluoroquinolonas pueden estar indicadas en infecciones urinarias de leves a moderadas, gonorrea no complicada, infecciones intestinales, infecciones óseas, sinusitis leve, bronquitis crónica, neumonía de la comunidad, cistitis y sinusitis aguda. La duración de la terapia depende del tipo y la gravedad de la infección y por lo menos debe continuarse por dos días después de desaparecido los síntomas.9
Estos antibióticos suelen tolerarse bien, pero en ocasiones pueden generar los siguientes efectos adversos sobre: a)SNC: psicosis aguda, agitación, alucinaciones, mareos, cefalea, nerviosismo, somnolencia e insomnio; b)Sistema digestivo: náusea leve, vómito o molestias abdominales, (todas o alguna combinación) colitis pseudomembranosa de moderada a fatal, se ha reportado con casi todos los antibacterianos, inclusive con las fluoroquinolonas; c) Cartílago de conjunción: las fluoroquinolonas puede producir daño al cartílago de crecimiento y artropatía, por ello está contraindicado en pacientes menores de 18 años y en mujeres embarazadas.5,6,7,9
La administración de quinolonas junto a otros compuestos pueden inducir a interaccionespoco deseables, de las cuales podemos mencionar algunas:2,3,5,6,9
Los ácidos: nalidíxico, oxolínico, pipemídico y piromídico, administrados junto a compuestos alcalinos incrementan las concentraciones séricas y urinarias de las quinolonas.
El ácido nalidíxico junto a warfarina, producen mayor efecto anticoagulante y peligro de hemorragias.
Todas las quinolonas de segunda generación, con excepción del ofloxacino, inhiben el metabolismo de la teofilina, aumentando su concentración en plasma sanguíneo.
Todas las quinolonas administradas vía oral junto a metoclopramida aumentan la motilidad intestinal y disminuyen el tiempo de vaciado del estómago, por otro lado si se administran con probenecid reducen la excreción de las mismas y extienden su semivida; y si se indicanjunto a hidróxido de aluminio o magnesio, estos reducen la absorción de quinolonas.
CARBAPENEMS
Considerados como los antibióticos de mayor espectro antibacteriano, los carbapenems se desarrollaron inicialmente para tratar microorganismos Gram-negativos productores de β-lactamasas y resistentes a penicilinas.2,4,6,10
ESTRUCTURA QUIMICA, CLASIFICACION Y ESPECTRO ANTIBACTERIANO
La obtención de la tienamicina, sustancia producida por Streptomyces cattleya, ha abierto la puertas para el desarrollo de los carbapenems. Inicialmente, debido a su inestabilidad química, se produjo un derivado estable el N-formimidoiltienamicida o imipinem, que administrado junto a la cilastatina fue el primer carbapenem disponible.4,11
El anillo carbapenem es un azobiciclo formado por la condensación de un anillo β-lactámico y otro pirrolidínico de 5 miembros e insaturado. Posee en posición 1 un átomo de carbono (carba) y un enlace no saturado entre 2 y 3 (em). Todos tienen en posición 6 un grupo hidroxietilo en configuración trans que protege al anillo β-lactámico de muchas serino-β-lactamasas y en posición 3 un radical carboxilo, importante para que el anillo pirrolidínico active al β-lactámico.11
Los distintos carbapenems son fruto de sustituciones en 1 y 2, de manera que se han creado principalmente y se hallan disponibles, cuatro miembros de este grupo antibiótico:11
Imipenem aprobado por la E.M.E.A., (European Medicines Agency, Agencia Europea del Medicamento)en 1985.
Meropenem E.M.E.A. 1994.
Ertapenem E.M.E.A. 2001.
Doripenem, certificado en julio de 2008 por la E.M.E.A.
Los carbapenems son estables frente a la mayor parte de las β-lactamasas y activos frente a estreptococos, estafilococos, enterobacterias, Escherichia coli, Klebsiella, Enterobacter, Proteus, Providencia, Salmonella, Shiguella, P. auruginosa, Haemophilus, Neisseria y bacterias anaerobias.1,10 Por otro lado, se ha propuesto una clasificación de carbapenems por espectro en tres grupos: a)Primer grupo: ertapenem, sin actividad frente a bacilos gramnegativos no fermentadores (BGNNF); b) Segundo grupo: imipenem, meropenem y doripenem, con actividad frente a BGNNF; b) Tercer grupo: carbapenems, aún no comercializados, con actividad frente a estafilococos resistentes a meticilina.11
FARMACODINAMIAY FARMACOCINETICA
Los carbapenems son antibióticos bactericidas que actúan inhibiendo la síntesis de la pared celular durante la transpeptidación, uniéndose a la serina residual de PBP (penicillin binding protein)situadas en la cara externa de la membrana citoplasmática, de modo que la pared celular se debilita y la bacteria muere por lisis. Frente a Listeria monocytogenes meropenem y ertapenem se comportan como bacteriostáticos, aunque la actividad intracelular de meropenem es bactericida a las 24 horas, de modo similar en infecciones por Streptococcus faecalis, el imipinem como otros antibióticos β-lactámicos, también es bacteriostático.1,11
Este grupo de antibióticos no se absorbe tras su administración oral, de manera que la vías intravenosa e intramuscular son las únicas, en su mayoría, penetran muy bien en la mayor parte de los fluidos y tejidos corporales (líquido peritoneal, bilis, orina, líquido sinovial, tejidos dérmicos, hueso, esputo, etc.) y se eliminan por orina, aunque también pueden eliminarse por hemodiálisis (cuadro N° 3). El imipinem se medica junto a la cilastatina puesto que al alcanzar niveles renales,ésta última impide su inactivación por la dehidropeptidasa-I renal.1,4,11
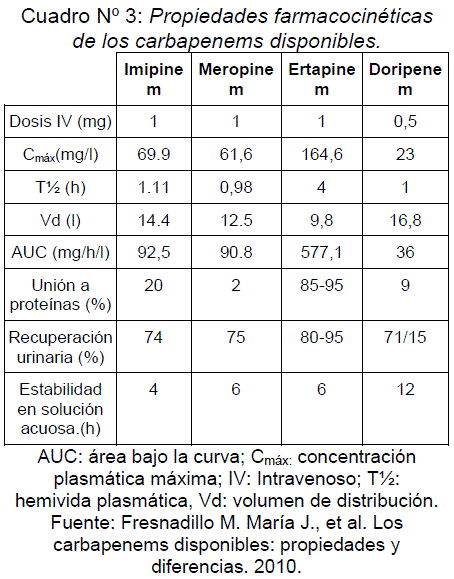
RESISTENCIA BACTERIANA
La resistencia a los carbapenems es poco común, es su mayoría los microorganismos que ofrecen esta baja resistencia son la P. aeruginosa, Acinetobacter spp. Klebsiella pneumoniae y S. Aureus resistente a meticilina. En Gram-negativos la resistencia, puede deberse a alteraciones en la permeabilidad, expulsión del antibiótico o inactivación por β-lactamasas, de manera contraria en Gram-positivos la resistencia se debe a modificaciones de las dianas.1,4
Con frecuencia la resistencia es cruzada, pero hay excepciones, como ocurre con cepas de P. aeruginosa que son sensibles a imipenem, resistentes a meropenem y doripenem, de Proteus-Morganella-Providencia o P. aeruginosa, resistentes a imipenem y sensibles a los otros carbapenems o de cepas de P. aeruginosa, Acinetobacter spp, Serratia marcescens, Enterobacter spp. y Citrobacter spp. resistentes a imipenem o meropenem y sensibles a doripenem.11
INDICACIONES TERAPEUTICAS, REACCIONES ADVERSAS E INTERACCIONES
Según Rang H.P. y colaboradores6 las principales indicaciones de los carbapenems incluyen infecciones de severas a complicadas causadas por microorganismos susceptibles, como P. aeruginosa, resistentes a otros medicamentos convencionales, a su vez están indicados en infecciones mixtas causadas por aerobios y anaerobios.6,11
De manera general los efectos adversos más comunes son: náuseas, convulsiones, vómito, diarrea, mareos, agranulocitosis, leucopenia, prurito, erupciones cutáneas, exantema, etc.5,9 La asociación de imipinem a encefalopatías ha sido discutido en los últimos años, pero constatar dicho cuadro todavía requiere de más estudios.12
Se debe evitar el uso de simultáneo con los siguientes medicamentos5,11
Probenecid: inhibe la secreción renal del carbapenem.
Ciclosporinas: pueden aumentar los efectos sobre SNC de ambos fármacos.
Ganciclovir: puede producir crisis convulsivas.
NOTAS
1 Univ. Tercer Año Facultad de Odontología UMSA
BIBLIOGRAFIA
1. Patiño Mendoza N.. Farmacología Médica. México D.F. Médica Panamericana. 2008:592-595; 616-619 [ Links ]
2. Velásquez P. L.. Farmacología Básica y Clínica. 17a ed. Buenos Aires. Médica Panamericana. 2004; 846-857 [ Links ]
3. Flórez J., Aramijo J. A., Mediavilla Á.. Farmacología humana. 5a ed. Madrid. ElsieverMasson. 2008;65:1265-1271. [ Links ]
4. Harman J.l G., Limbird L. E., Editor consultor GoodmanGilman A.. Las bases farmacológicas de la terapéutica. 10a ed. México D.F.MacGraw Hill. 2003:44:1197-1198; 45:1231-1232. [ Links ]
5. Bertram G. Katzung. Farmacología Básica y Clínica. 10a ed. México D.F. Manual Moderno. 2007: 764; 792-796. [ Links ]
6. Rang H. P., Dale M.M., Ritter J.M., Moore P.K.Farmacología. 5a ed. Madrid. Elsiever.200:. 429. [ Links ]
7. Alós J. I..Quinolonas. Formación Médica continuada en atención primaria.2003. [acceso 13 de septiembre de 2012] 21(5). Disponible en: http://www.elsevier.es/es/revistas/enfermedades-infecciosas-microbiologia-clinica-28/quinolonas-13137066-formacion-medica-continuada-2009 [ Links ]
8. Bello S., Torres A..Neumococo y resistencia a quinolonas. Editoriales, Archivos de Bronconeumología. 2003 marzo.[acceso 17 de septiembre de 2012] 39(3). Disponible en: http://www.archbronconeumol.org/bronco/ctl_servlet?_f=40&ident=13044146 [ Links ]
9. Rodríguez P. C., Rodríguez P. A., Hernández S. D.. Farmacología clínica. México D.F. Editorial McGraw Hill Interamericana. 2005; 384-385; 429-431. [ Links ]
10. Page P., Curtis M. J., Sutter C., Walker M. J.A., Horfman B. B. Farmacología integrada Madrid. Editorial Harcourd Brace.1998: 429; 437 [ Links ]
11. Fresnadillo M. M. J., García G. M.a I., García S. E., García S. J. E. Los carbapenems disponibles: propiedades y diferencias. Enfermedades Infecciosas y Microbiología Clínica. 2010. [acceso 17 de septiembre de 2012] 28(Supl.2) Disponible en: http://www.elsevier.es/sites/default/fileselsevier/pdf/28/28v28nSupl.2a13188595pdf001.pdf [ Links ]
12. Finkelsztejn A., Cabral L., Bragatti J.A.,Vitali da Silva A., Schumacher A. F. Imipenem-associated encephalopathy. ArqNeuropsiquiatr. 2010[acceso 17 de septiembre de 2012]68(1). Disponible en: http://www.scielo.br/pdf/anp/v68n1/a30v68n1.pdf [ Links ]