








Serviços Personalizados
Artigo
 Artigo em PDF
Artigo em PDF Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
Links relacionados
 Citado por SciELO
Citado por SciELO
 Similares em SciELO
Similares em SciELO
Bookmark
Revista SCientifica
versão impressa ISSN 1813-0054
SCIENTIFICA v.12 n.1 La Paz 2014
CASO CLINICO
Enfermedad De Creutzfeldt-Jakob: Reporte de un Caso
Creutzfeldt- Jakob Disease: Report Of One Case
Valentina Reyes H.1 Teresa Muñoz N. 2 Leonel Hidalgo A.3
1 Estudiante de Medicina. Facultad de Medicina, Universidad Católica de la Santísima Concepción
2 Interno de Medicina. Facultad de Medicina, Universidad de Concepción
3 Médico Cirujano, EDF, Hospital Hornopirén
Correspondencia a:
Valentina Reyes H.1
E-Mail:
RESUMEN
INTRODUCCIÓN: La enfermedad de Creutzfeldt-Jakob forma parte de un grupo de enfermedades conocidas como encefalopatías espongiformes transmisible o enfermedades priónicas. Clasificada en esporádica, familiar e iatro-génica, se manifiesta por cuadro demencial suba-gudo, síntomas motores, visuales, y mioclonías. Presentación del caso: Hombre, 42 años sin antecedentes mórbidos, presenta cuadro de dos meses de evolución caracterizado por mareos, desequilibrios y dificultad en la marcha. En extra-sistema neurólogo solicitó RM cerebro la cual mostró lesiones hiperintensas en ambos estriados principalmente a derecha e hiperseñal cortical a nivel frontal derecho y occipital bilateral. Examen físico general: Sin alteraciones. Examen neuroló-gico: Vigil, consciente, atento, orientado témporo espacialmente, de buen humor; memoria a largo y corto plazo conservadas; funciones encefálicas superiores conservadas ; examen de pares craneales, diplopía mayor a izquierda sin otra alteración; Examen motor, tono normal, trofismo conservado, fuerza conservada; marcha atáxica, dismetría izquierda; sin alteraciones sensitivas. Antecedentes familiar: hermano fallecido por cuadro similar. Dada la clínica se sospecha enfermedad de Creutzfeldt-Jakob o una encefalopatía tóxico me-tabólica inespecífica, se solicita un Electroencefalograma y una Punción lumbar ara pro teína 14-3-3 en espera de resultados. Actualmente se encuentra con Clorpromazina 25 mg c/noche vo y Paracetamol 500 mg vo SOS. Discusión: El diagnóstico de ECJ se sospecha con la clínica, y se fundamenta con hallazgos característicos en RM, EEG y análisis de LCR. Ante la sospecha de la forma familiar se sugiere estudio genético. Al ser una enfermedad invariablemente mortal y sin tratamiento, dificulta la decisión entre realización de estudios e intervenciones, contra el manejo expectante.
Palabras claves
Enfermedad de Creutzfeldt-Jakob, Encefalopatías espongiformes transmisibles, Enfermedades por prión
ABSTRACT
Introduction: Creutzfeldt-Jakob disease belongs to a group of diseases known as transmissible spongiform encephalopathies or prion diseases. Classified in sporadic, familial and iatrogenic, manifested by subacute dementing box, motor symptoms, visual, and myoclonus Case Presentation: male, 42 years without morbid history, presents picture of two months' duration characterized by dizziness, imbalance and difficulty in underway. In extra-sistema neurologist asked which brain MRI showed hyperintense lesions in both striated mainly cortical hyperintense to right and right frontal and occipital bilaterally. Physical Exam General: No changes. Neurological examina-tion: Vigil, aware, attentive, temporomandibu-lar spatially oriented, good-humored; long memory and short-term preserved; higher brain functions preserved; examination of cranial nerves, greater left diplopia without other alteration; Consideration motor normal tone, preserved trofismo, strength preserved; ataxic gait, dysmetria left; no sensory disturb-ances. Family Background: brother died from similar symptoms. Given the Creutzfeldt-Jakob disease clinic suspect a nonspecific metabolic or toxic encephalopathy, requested an Electro-encephalogram and 14-3-3 lumbar puncture awaiting results. Currently with Chlorproma-zine 25 mg c / vo night and Paracetamol 500 mg po SOS. Discussion: The diagnosis of Creutzfeldt-Jakob disease is suspected with the clinic, and is based with characteristic MRI findings, EEG and CSF analysis. Suspecting the familiar form genetic study suggests. As an invariably fatal disease without treatment, difficult decisión between studies and interven-tions with expectant management
Keywords
Creutzfeldt-Jakob disease, transmissible spongiform encephalopathies, prion diseases
INTRODUCCIÓN
La enfermedad de Creutzfeldt-Jakob (ECJ) es una enfermedad priónica neurodegenerativa que afecta el SNC del hombre, transmisible e invariablemente mortal1'2. Hay 3 categorías principales de la ECJ: esporádica, hereditaria y adquirida3, siendo la forma esporádica la más frecuente a nivel mundial con aproximadamente 90% de los casos4; sin embargo, en Chile la forma familiar se encuentra en mayor medida 5,6. La característica principal de este trastorno es la alteración de una proteína celular ubicada en la membrana celular y la sinapsis (PrPc) por una proteína insoluble (PrPsc) que se acumula progresivamente con la evolución de la enfermedad en forma de placas amiloides extracelu-lares7.
El diagnóstico de una probable ECJ se basa en los hallazgos clínicos como inestabilidad de la marcha, temblores, deterioro cognitivo e intelectual, cambios de personalidad y parálisis, en combinación con los resultados de una serie de pruebas complementarias como el electroencefalograma (EEG), el estudio de líquido cefalorraquídeo (LCR) o la neuroimagen. El único método de diagnóstico es el estudio anatomopa-tológico del cerebro8. Damos a conocer un caso recogido este año en el HCHM con una forma de presentación y evolución clásica.
PRESENTACIÓN DEL CASO
Paciente de 42 años, sexo masculino, sin antecedentes mórbidos, con historia familiar que destaca fallecimiento de hermano por una posible ECJ. Ingresa desde el Consultorio Adosado de Especialidades (CAE) donde consultó por cuadro de 1 mes de evolución caracterizado por ataxia progresiva, bradipsiquia y diplopía, TAC cerebro normal, RNM sugiere considerar ECJ o Encefalopatía Tóxico Metabólica inespecífica por lo que se hospitaliza para estudio. Se realiza EEG y PL. Se mantiene estable con disartria, diplopía, dismetría y adiadococinesia mayor a izquierda. Se decide alta médica y continuar estudio de forma ambulatoria. Es evaluada en extrasistema, donde se solicita RM cerebro la cual mostró lesiones hiperintensas en ambos estriados principalmente a derecha e hipersefial cortical a nivel frontal derecho y occipital bilateral, dado estos antecedentes. Se decide su hospitalización en Servicio de Neurología de HCHM para estudio. A su ingreso, presenta examen físico general sin alteraciones, examen neurológico, vigilia consciente, atento, orientado témporo espacialmente, Glasgow 15, de buen humor; memoria a largo y corto plazo conservadas, bradipsíquico, funciones encefálicas superiores conservadas. Al examen de pares craneales, diplopía mayor a izquierda sin otra alteración. Examen motor, tono normal, trofismo conservado, fuerza conservada; marcha atáxica, dismetría izquierda; sin alteraciones sensitivas. Signos meníngeos negativos. Dada la clínica se sospecha un ECJ o una encefalopatía tóxico metabólica inespecífica, se solicita un electroencefalograma (EEG) y una punción lumbar (PL) para proteína 14-3-3.
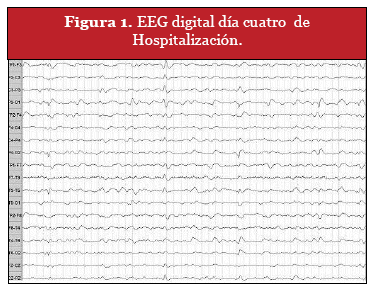
Durante su estadía, se mantiene estable en su condición, con buena hemodinamia, PA: 120/70 mmHg, Fc:99 x', SatO2:93% , FiO2:o,2l, T°:36,4 °C.
Al examen físico, vígil, orientado temporo espa-cialmente, disártrico. Cardiopulmonar: Ritmo regular en 2 tiempos sin soplos, Murmullo pulmonar presente, sin ruidos agregados. Abdomen, blando, depresible, indoloro, sin masas palpables. Extremidades, sin edema y sin signos de TVP. Al examen neurológico, Glasgow 15, diplopía mayor a izquierda, marcha atáxica, dismetría izquierda, sin alteraciones sensitivas.
Se mantiene con reposo relativo, régimen común, Clorpromazina i comprimido 25 mg noche cada noche vía oral, Paracetamol 500 mg, vía oral SOS a la espera del informe EEG y realización Punción lumbar para proteínas 14-33.
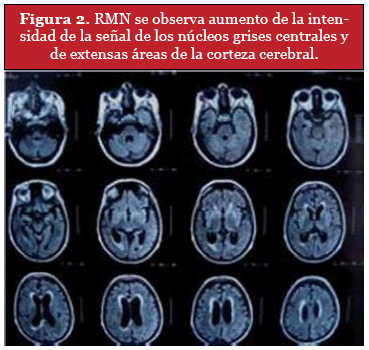
DISCUSIÓN
Se estima que en nuestro país cada año fallecen 40 personas por la ECJ, y que Chile muestra la más alta incidencia a nivel mundial de la forma familiar de la ECJ9 Se presenta un caso con una sintomatología inicial y un curso evolutivo clásico, lo que remarca la importancia no solo de los criterios diagnósticos de la ECJ10, sino también de la sospecha diagnóstica en sí misma.
La ECJ afecta al sistema nervioso, y sus síntomas iniciales son la demencia, alteraciones de personalidad, deterioro de memoria, juicio y pensamiento, sumado a problemas de coordinación muscular. Los afectados pueden presentar insomnio o depresión11.
Frente a la sospecha de ECJ, es necesario descartar otras formas tratables de demencia, como encefalitis o meningitis crónica, ya que la ECJ no causa fiebre ni compromiso del estado general, razón por la cual es necesario un examen neurológico realizado por especialistas11.
Un 10% de los casos de ECJ son de causa genética. La clasificación etiológica de los casos es importante por motivos epidemiológicos y para realizar un asesoramiento de riesgo genético correcto a los familiares de un afecto. Sin embargo, se sabe que hasta un tercio de los casos de ECJ genética se presentan como esporádicos por la ausencia de antecedentes familiares y las características clínicas tampoco permiten diferenciar un caso esporádico de uno genético. Este caso refuerza, pues, la idea de que el estudio genético se debe realizar en todo caso de ECJ con independencia de la historia familiar y más en la provincia de Nuble, donde la incidencia es elevada, ya que entre los años 2006 y 2010 se presentan anteceden-ites de 13 casos, dentro de los cuales existen dos pacientes hermanas diagnosticadas. Nuestro país posee elevada frecuencia de casos familiares de ECJ12.Esto amerita tener una alta sospecha y recabar antecedentes familiares oportunamente; sin embargo, en el HCHM no se realiza un estudio genético. Es por esto que creemos de vital importancia y considerando que el HCHM es el centro de referencia de la provincia del Nuble, que debería contar con esto. Dada la importancia epidemiológica y sobre la salud humana y animal, es obligación de todos los trabajadores de salud estar atentos a este diagnóstico
REFERENCIAS
1) Edgeworth JA, Farmer M, Sicilia A, Tavares P, Beck J, Campbell T, et al. Detection of prion infection in variant Creutzfeldt-Jakob disease: a blood-based assay. Lancet 2011; 377 (9764): 487-93- [ Links ]
2) Gambetti P, Kong Q, Zou W, Parchi P, Chen SG. Sporadic and familial CJD: clas-isification and characterization. Br Med Bull 2003; 66: 213-239. [ Links ]
3) National Institute of Neurological Disorders and Stroke (NINDS) [Internet]. Maryland: National Institutes of Health (NIH) [actualizado 16 May 2012; citado 9 Oct 2012]. Disponible en: http://www.ninds.nih.gov/disorders/cjd/detailcjd.htm. [ Links ]
4) Gálvez S, Cartier L. Análisis clínico de una serie de 69 casos definitivos de Enfermedad de Creutzfeldt-Jakob ocurridos en Chile entre 1960 y 1985. Rev Med Chil 1987; 115 (12): 1148-54. [ Links ]
5) Ministerio de Salud. Vigilancia Epidemiológica de Enfermedad de Creutzfelt- Jakob en Chile. 2007. Disponible en: epi.min-sal.cl/epi/html/normas/circul/Circu-ilar-ECJ.pdf [Consultado el 25 de mayo de 2011]. [ Links ]
6) Hernández FJ, Martínez M, Esteban M, Palacios S, Jensen F. A propósito de la incidencia de la Enfermedad de Creutzfel-idt- Jakob en el área de salud de Lanzaro-ite. Descripción de 2 casos definitivos. Neurologia 2005; 20 (1): 41-4. [ Links ]
7) PRUSINER SB. Neurodegenerative disease and prions. N Engl J Med 20015344:1516-26. [ Links ]
8) Creutzfeld Jackob Disease Foundation [Internet]. Ohio: The CJD Foundation; 2012 [citado 9 Oct 2012]. Disponible en: http://www.cj dfoundation.org/webfm_send/13 [ Links ]
9) Gajdusek DC. Infectious Amyloids: Subacute Spongiform Encephalopathies as Transmisible Cerebral Amyloidoses. In: Fields BN, Knipe DM, Howley PM y cois, ed. Fields Virology. Filadelfia: Editorial Lippincott-Raven Publish-ers, 1996; 2851-900. [ Links ]
10) Mosqueira AJ, Barroso A, López-Manzanares L, Canneti B, Zapata G, Palmí I, et al. Falsa enfermedad de Creutzfeldt-Jakob [abs-tract]. RevNeurol 2011; 52: 567. [ Links ]
11) La enfermedad de Creutzfeldt Jakob es más frecuente en Chile que en el resto del mundo [en línea] Julio 6 2004 [fecha de acceso 10 de Julio]. URL disponible en http://www. capp.uchile.cl/undin2/actuales /notÍ4OOi.shtml
12) Morgado-Linares RY, Ruiz-Pefia JL, Páramo MD, Díaz-Delgado M, Izquierdo G. Características clínicas de la enfer-imedad de Creutzfeldt-Jakob familiar y mutación E200K en España. RevNeurol 2007; 44 (3): 150-153. [ Links ]