








Serviços Personalizados
Artigo
 Artigo em PDF
Artigo em PDF Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
Links relacionados
 Citado por SciELO
Citado por SciELO
 Similares em SciELO
Similares em SciELO
Bookmark
Revista SCientifica
versão impressa ISSN 1813-0054
SCIENTIFICA v.12 n.1 La Paz 2014
ARTÍCULO DE REVISIÓN
Análisis del gen hsd17b10: un ejemplo de regulación epigenética por metilación de segmentos cpg y su relación con patologías ligadas al cromosoma x
Analysis of hsd17b10 gene: an example of epigenetic regulation by methylation of cpg segments and its relationship with x-linked pathologies
Del Barco Gómez Sergio1 Encinas Vargas Dorian Victor1 Eguino Arenas José Manuel1 De la Torre Urey Cesar Alejandro1
1 Estudiante Tercer año Facultad de Medicina UMSA
Correspondencia a: Encinas Vargas Dorian Victor E-mail: dorianencinas@gmail.com
RESUMEN
El gen HSD17B10 se encuentra en Xp 11.2 y codifica la enzima HSDlO. Este gen ha sido reportado como uno de los pocos genes que escapan a la inactivación del cromosoma X. Este gen presenta abundantes regiones de CpG (Citosina-fosfato-Guanina) en su estructura molecular. Los dinu-cleótidos pertenecientes a los segmentos CpG son metilados en posición 5 de la citosina, este proceso se encuentra asociado a la represión transcrip-cional y a la inactivación de la conformación de la cromatina.
HSDlO es una enzima multifuncional que cataliza procesos importantes tales como el catabolismo de la isoleucina, la oxidación de la alopregneno-lona en 5a dihidroprogesterona. Además se ha demostrado que esta enzima se puede unir al péptido amiloide P y el receptor de estrógeno alfa. Es a razón de todas estas funciones que la deficiencia de esta enzima se ve traducida en alteraciones diversas de la función mental propias de la alteración del metabolismo de los aminoácidos, además de estar relacionado a otras patologías como son la enfermedad de Alzheimer y la enfermedad de Parkinson. El cromosoma X inactivo (Xi) por metilación presenta genes capaces de escapar a la inactivación. La hipo metilación de los CpG correlacionado con la expresión de genes pertenecientes a Xi da lugar a una serie de patologías clínicas pudiéndose mencionar la deficiencia de la producción de la enzima y enfermedades congénitas ligadas a X.
El objetivo de esta revisión es resaltar la importancia de la metilación de segmentos CpG como un mecanismo de regulación epigenética.
Palabras clave Cromosoma X, Metila-ción del DNA, Represión epigenética, silencia-miento de genes
ABSTRACT
HSD17B10 gene is located in Xp 11.2 and codi-fies the HSDlO enzyme.lt has been reported as one of the few genes that escapes the inactiva-tion of X chromosome. This gene presents a lot of CpG regions (Cytosine - phosphate - Guani-dine) in its molecular structure. Dinucleo-tidsbelonging to the CpG segments are methyl-ated in 5 position of the cytosine; this process is associated to the transcriptional repression and to the inactivation of chromatin conformation. HSDlO is a multifunctional enzyme that cata-lyzes important processes such as the isoleu-cine catabolism, oxidation of allopregnenolone in to sadihydroprogesterone. Moreover, it has been demonstrated that this enzyme can be joined to the amyloid peptideP and the alpha estrogen receptor. As a result of these func-tions, the deficiency of this enzyme its translat-ed into diverse alterations of mental function which are common in the alteration of amino-acids metabolism. Furthermore, this has been related to other pathologies like Alzheimer and Parkinson diseases.
The X chromosome inactivated by methylation (Xi) presents genes capable of escaping to the inactivation. The CpGhypomethylation, corre-lated to the expression of genes belonging to Xi,gives raise to a series of clinical pathologies such as the deficiency of the enzyme production and other X-linked pathologies. The objective of this revisión is to highlight the importance of the methylation of CpG segments as a mechanism of epigenetic regulation.
Keywords X sexual chromosome, DNA methylation, Epige-netic repression, Gene silencing
INTRODUCCIÓN
El presente trabajo tiene como fundamento señalar la importancia del grado de metilación del gen HSD17B10 en sus segmentos CpG que está relacionado con la inactivación de la expresión de secuencias procedentes del cromosoma X, además de estar vinculado a patologías presentes en el ser humano. Este es un tópico de gran trascendencia, debido a que todo el conjunto de mecanismos que produce la inactivación del cromosoma X constituyen una ejemplar demostración de los mecanismos de regulación epigenética, uno de los últimos avances de la biología molecular que amplió nuestra visión de la regulación de la expresión genética más allá del dogma molecular, aún todavía incomprendido. Para esto, siguiendo un modelo deductivo, se tomó en cuenta al gen HSD17B10, que codifica la enzima HSD10, viendo la importancia de su mecanismo de regulación en el desarrollo de patologías por deficiencia de la enzima.
DESARROLLO DEL TEMA
Funciones de la HSD10
La HDS10 es una enzima multifuncional que al principio sólo se le conocía por el papel que tiene en el catabolismo de la isoleucina, pero con la aparición de diferentes y nuevos estudios se han encontrado nuevas y variadas funciones de la misma, que explican todas las patologías relacionadas a su deficiencia1'4.
La enzima actúa en el metabolismo de la isoleucina, específicamente en la oxidación de la 2-metil 3-hidroxibutiril CoA en 2-metilacetoacetil CoA, con la participación de un agente reductor como ser el Nicotin Adenin Dinucleótido (NAD) en el catabolismo de la isoleucina. La deficiencia en esta enzima conlleva la acumulación de los productos ya formados, es decir la tiglil CoA y la misma 2-metil 3-hidroxibutiril-CoA5,6.
Además la HSD10 cataliza la oxidación de la alopregnenolona en 5a dihidroprogesterona, con ayuda del NAD como coenzima, con su correspondiente inactivación4'5. La alopregnenolona radica su importancia debido a que se une a receptores de GABA, estimulando su función de neurotransmisor inhibidor7,8.
Se ha demostrado que su actividad se puede inhibir cuando se une a determinadas moléculas como ser el péptido amiloide-P o el receptor de estrógeno alfa. En el primer caso, esa unión está muy relacionada a la disfunción mitocondrial que se sufre en la enfermedad de Alzheimer, al tener niveles acumulados de la enzima a nivel de hipocampo. También ha sido relacionada en los mecanismos de patogenicidad de la enfermedad de Parkinson, al controlar y regular las neuronas que contienen dopamina9,11.
Análisis epigenético del gen HSD17B10
La enzima HSD10 esta codificada por el gen HSD17B10. Este gen se encuentra en el cromosoma X, específicamente fue mapeado en la región Xpn.2, junto a otros genes como el HUWEi que codifica la ligasa de ubiquitina E3, el gen SMCiA y RIBC11. Este es un aspecto muy importante, ya que dicho gen ha sido reportado como uno de los que escapa a la inactivación del cromosoma X. En estudios de este cromosoma se ha encontrado que la mayoría de los genes que escapan a la inactivación de X se encuentran en el brazo pequeño (p) del cromosoma X12'14.
El gen HSD17B10 ha sido altamente conservado en la evolución filogenética de las especies, ya que desde los insectos muestra la misma estructura básica, con algunas ligeras variaciones en el número de exones, intrones y aminoácidos13.
El gen HSD17B10 (antes llamado HADH2), contiene en su estructura seis exones y cinco intrones, además de codificar la enzima HSD10 de 261 aminoácidos. Esta es una molécula homote-tramérica, con un centro de 7 hojas P paralelas rodeadas de seis hélices a, estas últimas implicadas en la interacción con la coenzima NAD3.
Realizando el análisis del gen se encontró que en su región 5' terminal tiene tres sitios de inicios de transcripción, los más importantes ubicados a -37 y -6 bases corriente arriba del codón de inicio ATG y uno más a -12. Además se encontró en esta misma región una isla de CpG, conteniendo desde -171 a -6 bases corriente arriba del codón de inicio, con una cantidad de 23 dinucleótidos CpG. Estos dinucleótidos están presentes en gran cantidad en los genes "house-keeping", además de ser susceptible a sufrir metilación. Otro aspecto interesante de este genes que no contiene una caja promotora con la secuencia TATA, sino que está remplazado por una caja CCAAT, ubicado de -52 a -56 bases corriente arriba del codón de inicio7.
Rol de la metilación de las islas CpG en la inactivación del cromosoma X.
Es preciso aclarar acerca de la inactivación del cromosoma X, ya que este proceso está muy relacionado a la expresión de dicho gen.
Puesto que las mujeres contienen una carga genética con dos cromosomas X, es necesario que uno de estos cromosomas sea inactivado; para este fin deben activarse diversos mecanismos epigenéticos para la inactivación del cromosoma X, mecanismos que son similares y se repiten en los demás procesos de regulación de expresión genética. Es así que las mujeres tienen un cromosoma X activo (Xa) y un cromosoma X inactivo (Xi)- Esta observación es la que hizo la investigadora Mary Lyon en 1961. A partir del descubrimiento del corpúsculo de Barr en los neutrófilos y las apreciaciones de Susuma Ohno que dedujo que éste estaba relacionado a una parte inactiva de la cromatina, fue Lyon quien propuso que había una inactivación aleatoria de uno de los cromosomas X, proceso llamado en su honor como lionización5.
Para que se produzca la inactivación de este cromosoma es necesaria en primer lugar la participación de un locus en el cromosoma X llamado Xic, ubicado en la región Xqi3, que contiene cuatro genes, de los cuales el principal para la inactivación es el gen Xist. Es entonces que en las blastómeras del embrión femenino se debe producir la inactivación al azar de uno de los cromosomas X, dando como resultado un cromosoma Xa que silencia al gen Xist y el otro cromosoma Xi que da lugar a la expresión de este mismo gen. Existen muchos trabajos que hablan de las diferentes condiciones que hacen que este gen se exprese o no. Entre las ideas más aceptadas se hablan de diferentes tipos de promotores en los cromosomas, de una hipera-cetilación de la histona H4 solo visto en células madres de genotipo femenino, etc. En una de las teorías más interesantes se menciona la existencia de un factor que llega a proteger únicamente a uno de los genes Xist para su expresión, mientras que el otro se queda desprotegido, y por ende, inactivado. Además el gen Tsix,antagonista de Xist, puede ser importante en la lionización, ya que este sólo se expresa en el cromosoma X activo. Sin embargo lo que ha quedado establecido es que la activación del gen Xist es imprescindible para los demás cambios epigenéticos que posibilitan el silenciamiento del cromosoma X1,18.
En estudios realizados en mamíferos se ha visto que es preferentemente el cromosoma X paterno el que es inactivado. Esto puede deberse a que el cromosoma inactivado muestra una variedad especial de histona, la histona macro H2A. Según algunos autores esta histona estaría ligada ya al cromosoma paterno desde el mismo proceso de la meiosis masculina. Dicha histona presenta mucha analogía con la histona H2A, con la diferencia que en su extremo C-terminal posee un dominio macro, de una secuencia de aminoácidos cuyo mecanismo de acción en la represión de la expresión del gen aún no ha sido develado5.
Según investigaciones realizadas y acorde con la investigadora Mary Lyon, se indica que la inactivación del cromosoma X estaría fuertemente ligada a la presencia de una serie de secuencias denominadas LINE (Long Interspersed Nuclear Elements), dado que los cromosomas X de ratones y humanos contienen abundante cantidad de LINE-11'15'18'20.
Sin embargo, el proceso de inactivación de X no llega a abarcar todo el cromosoma, sino que un 15% de los genes expresados por el mismo cromosoma escapan a la inactivación de X. Estos genes están ubicados principalmente en el brazo corto del cromosoma X (Xp). Así mismo, es interesante observar la relación entre la cantidad de secuencias LINE en las partes del cromosoma, las mismas que se encuentran agrupadas principalmente alrededor del gen Xist, a la vez de ser más escasas en Xp, especialmente en la región Xp22, siendo demostrado ser la región con la mayor cantidad de genes que escapan a la inactivación de X5.
Todos estos procesos serían inútiles sin los adecuados procesos epigenéticos que logren asegurar la inactivación de los genes del cromosoma X. Estos mecanismos son los mismos que sirven para regular la expresión del DNA en los demás cromosomas. Sin embargo, los que más se repiten en el cromosoma X son la regulación de las
histonas, ya que pueden aparecer historias hi-peracetiladas, como la historia 4, o aparecer un tipo especial de histona, como la histona macro H2A, y otro tipo de histonas especiales como la H3K27me3 y H3K9me3 (Figura 1). No obstante, uno de los mecanismos descubiertos sobre la represión de expresión del DNA que revolucionó el estudio de la biología molecular fue la capacidad de metilación del DNA en los residuos de citosina, para su inactivación. Esta inactivación se da principalmente en las llamadas islas CpG17.
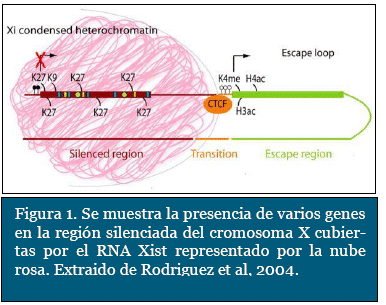
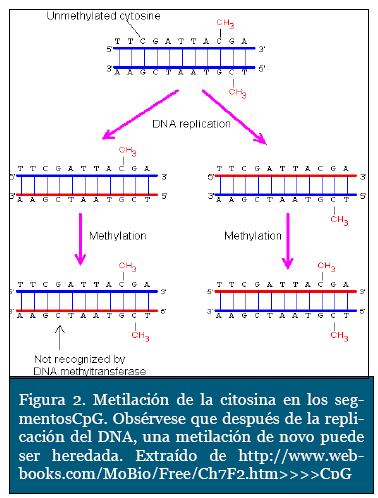
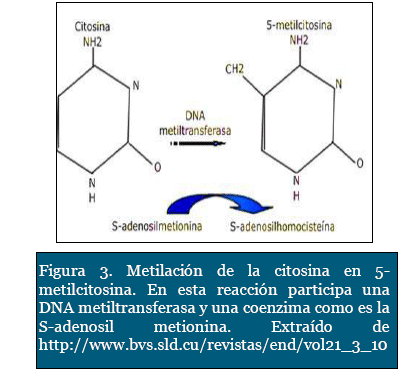
Las islas CpG son pequeños segmentos del ge-noma ricos endinucleótido citosina - guanina ubicadas en la región 5' de la mitad del total de los genes humanos. Éste proceso de metilación ocurre en el carbono 5 del anillo pirímidinico de la citosina, cuya reacción es catalizada por ciertas enzimas denominadas ADN-metil transfera-sas, con la ayuda de una coenzima transportadora de grupos metilo, como es la S-adenosilmetionina (Figura 3). Un aspecto interesante de este mecanismo, es que, después de producida una metilación de novo, posteriormente es heredada para las siguientes copias del genoma (Figura 2). Estas islas CpG están localizadas preferente y estratégicamente sobre los promotores de la transcripción de los genes. La metilación anormal de estas islas está a menudo relacionada con procesos neoplásicos, ya que puede ocasionar la falta de expresión de genes antitumorales. Así mismo, ha sido asociado con otras patologías ligadas al cromosoma X como el síndrome de X frágil7.
En rol que cumplen las islas CpG en la inactivación del cromosoma X, los genes del cromosoma X activo mostrarán sus regiones CpG poco metiladas. En cambio los genes del cromosoma X inactivo mostrarán un alto grado de metilación de sus regiones CpG, llegando casi al 100%, exceptuando aquellas regiones que contengan los genes que escapen a la inactivación de X.
Por lo tanto, si hacemos el recuento de metila-ción de un gen en sus dos alelos, haciendo un promedio entre el o% de metilación de uno y el 100% del otro, nos muestra una metilación promedio del gen de un 50%, que sería el estándar para definir un gen que este inactivo. Los genes del cromosoma X del genotipo masculino muestran un nivel cercano a o % de metilación13.
A pesar de la gran relación existente entre la inactivación de los genes del cromosoma X y la presencia de los dinucleótidos CpG, no se encontró ninguna relación entre la distribución de los locus de los genes que se inactivan con la ubicación de las islas CpG5.
En el caso del gen HSD17B10 se demostró que de los 23 dinucleótidos CpG encontrados en el extremo 5' del gen, los varones contaban con menos del 3% metilados, como se esperaba. En el caso del genotipo femenino ninguna de las regiones alcanzo más del 30% de metilación de las citosinas de las islas CpG. Comparando estos datos con el de los genes sometidos a inactivación, se presume que el gen HSD17B10 estaría comprendido dentro del lote de genes que escapa a la inactivación de X. (Figura 4)7.

Al darse la expresión de este gen en ambos cromosomas, se estaría hablando de una expresión bialélica, lo que daría oportunidad a mujeres con este gen mutado a que contengan células en las cuales este gen se pueda expresar en algunas células y así su traducción produzca un poco de la enzima necesaria15.
Esto implicaría que las mujeres portadoras del gen no serían sintomáticas, pero existen algunos estudios que muestran que estos genes sí mostrarían inactivación por X y que además no sería aleatoria, por lo que algunas células pueden inactivar al gen no afectado y así desarrollar cierto grado de sintomatología de deficiencia de la enzima. Sin embargo, estos estudios carecen de validez porque no se realiza un análisis epi-genético de los mecanismos de regulación de expresión genética12.
DISCUSIÓN
Las mutaciones que afectan al gen HSD17B10 se caracterizan por ser mutaciones de transversión en las que el cambio de bases afecta la estructura molecular de la enzima HSD10 dando origen a patologías relacionadas con el metabolismo de la isoleucina y otros ácidos grasos metilados, además de neuroesteroides como la alopreg-nenolona. Si bien durante el metabolismo de la isoleucina ya se habían descrito otros factores que al fallar ocasionaban datos laboratoriales similares a los producidos por la falla de la enzima se sostiene que estas causas ajenas a la mutación del gen no presentan sintomatología aparente.
Se destaca la multifuncionalidad de la enzima HSD10 en diversos procesos metabólicos a nivel encefálico como un importante regulador de la trasmisión de señales nerviosas mediante su mecanismo de unión a los receptores de GABA, al mismo tiempo que se encuentra implicado en ciertas patologías como la enfermedad de Par-kinson y la enfermedad de Alzheimer en las cuales su rol todavía es desconocido.
La metilación de los segmentos CpG cumple un importante papel como un mecanismo más de regulación de expresión genética del DNA, especialmente en el caso particular de la inactivación de genes encontrados en el cromosoma X. Sin embargo existen genes que escapan a la inactivación por la metilación no estando relacionados con CpG. Pudiendo estos ser causantes de múltiples patologías congénitas especialmente en varones siendo que en estos el gen X tiene una expresión monoalélica.
Se resalta la importancia que tiene el conocimiento de éste y otros mecanismos epigenéticos por parte de los médicos en formación, dado que la biología molecular presenta un camino aún bastante desconocido, pero que al dilucidarlo brindará la oportunidad de implementar un nuevo paradigma en el conocimiento médico. La epigenética abre las puertas a los profesionales en salud del futuro para no quedarse estancados en la medicina clásica y desarrollar tratamientos para diversas patologías a nivel molecular.
REFERENCIAS
1. Berletch J, Yang F, Xun J, Carrel L, Disteche CM. Genes that excape from X inactivation. Hum Genet.[online] 2011 Ago [acceso 18 de septiembre de 2013]; 130(2): [237-45]. Disponible en: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC313Ó2O9/pdf/nihms3O4O19. [ Links ]
2. Berletch J, Yang F, Xun J, Disteche CM. Escape from X inactivation in mice and humans. Genome Biol.[online] 2010 [acceso 18 de septiembre de 2013]; 11(6): [213]. Disponible en: http://link.springer.com/article/10.1007/s00439-011-1011-z [ Links ]
3. SeaverLH, He X, Abe K, Cowan T, Enn GM, Sweetman L et al. A novel mutation in the HSD17B10 of a 10-Year-Old Boy with Refractory Epilepsy. PlosOne [online] 2011 Nov. [acceso 18 de septiembre de 2013]; 6(11): [1-8] [ Links ]
4. Yang S, Dobkin C, He X, Phillip M, Brow Y. A 5-methylcytosine hotspot responsible for the prevalent HSD17B10 mutation. Gene [online]. 2013 [acceso 18 de agosto de 2014]; 515(12): [380-4]. Disponible en: http://0nlinelibrary.wiley.com/d0i/10.1002/9780470015902.a0005O27.pub2/abstract [ Links ]
5. Carrel L, Hungtington W. X-inactivation pro-file reveáis extensive variability in X-linked gene expression in females. Nature [online]. 2005 Mar [acceso 18 de septiembre de 2013]; 434(17): [400-4]. Disponible en: http://www.nature.com/nature [ Links ]
6. De Mello JC, De Araujo ES, Stabellini R, Fraga AM, De Souza JE, Sumita D et al. Random X inactivation and extensive mosaicism in human
placenta revealed by analysis of allele-specific gene expression along the X chromosome. PlosOne [online] 2010 [acceso 20 de septiembre de 2013]; 5(6). Disponible en: http://www.plosone.org/article/info%3Ado1%2Fio.1371%2Fjournal.pone.0010947
7. Froyen G, Corbett M, Vandewalle J, Jarvel I, Lawrence O, Meldrum C et al.. Submicroscopic Duplications of the Hydroxysteroid Dehydro-genase HSD17B10 and the E3 Ubiquitin Ligase HUWEi Are Associated with Mental Retarda-tion. Am J MedGenet [online]. 2008 febrero [acceso 18 de septiembre de 2013]; 82 [432-3]. Disponible en: http://www.ncbl.nlm.nih.gov/pubmed/18252223
8. Yang S, He X, Miller D, McMenamin J, Philipp M et al. Hydroxisteroid (17P) dehydrogenase X in human health and disease. Molecular and celular endocrinology [online]. 2011 [acceso 18 de septiembre de 2013]; 343(1): [1-6]. Disponible en:
http://www.sciencedirect.com/science/article/PÜ/S0303720711003194
9. Tresguerres J. Fisiología Humana. 4a Ed. McGraw Hill Interamericana; Madrid: 2011. Pp 134-6 [ Links ]
10. Yao J, Irwin RW, Zaho L, Nilsen J, Hamil-ton RT, Diaz Brinton R. Mitochondrial bioener-getics déficit precedes Alzheimer's pathology in female mouse model of Alzheimer's disease. PNAS [online]. August 25, 2009 [acceso 26 de agosto de 2013]; 106(34):14070-5. Disponible en:http://www.pnas.org/cgi/dol/10.1073/pnas.0903563106
11. Coskun P, Wyrembak J, Schriner SE, Chen HW, Marciniack C, LaFerla F et al. A mitochodrial etiology of Alzheimer and Parkinson disease. BiochimicaetBiophysicaActa (BBA) -General Subjects [online]. 2012 [acceso 15 de agosto de 2014]; 182o(s):553-64. Disponible en:http://www.sciencedirect.com/science/article/ PÜ/S0304416511002054
12. Garcia-Villoria J, Gorti L, Madrigal I, Fons C, Fernández C, Navarro-Sastre A et al. X-inactivation of HSD17B10 revealed by cDNA analysis in two female patients with 17-P-hidroxisteroid dehydrogenase 10 deficiency. EuropeanJournal of Human Genetics [online]. 2010 [acceso 18 de septiembre de 2013]; 18: [1353-5]- Disponible en http://www.nature.com/ejhg [ Links ]
13. Yang S, Dobkin C, He X, Brow Y. Transcrip-tion start sites and epigenetic analysis of tne HSD17B10 proximal promoter. BMCBiochemistry [online]. 2013 s.f. [acceso 18 de septiembre de 2013]; 14(17): [1-6]. Disponible en:
http://0nlinelibrary.wiley.com/dol/10.1002/9780470015902.a0005O27.pub2/abstract
14. Yang S, He X, Isaacs C, Dobkin C, Miller D, Phillip M. Roles of i7P-hydroxisteroid dehydrogenase type 10 in neurodegenerative desorders. The Jornal of steroid biochemistry and molecular biology [online]. 2014 [acceso 18 de agosto de 2014]; 143:460-72. Disponible en: http://www.sciencedirect.com/science/article/ PÜ/S0960076014001241
15. Yang S, He X, Olpin SE, Sutton VR, McMenamin J, Philipp M et al. Mental retarda-tion linked to mutations in the HSD17B10 gene interfering with neurosteroid and isoleucine metabolism. PNAS [online]. 2009 sep [acceso 18 de septiembre de 2013]; 106(25): [14820-4]. Disponible en: http://www.pnas.org_cgi_doi_io.iO73_pnas.0902377106
16. Zschocke J. HDS10 disease: clinical conse-quences of mutations in the HSD17B10 gene. Journal of inherited metabolic disease [online]. 2012 [acceso 18 de septiembre de 2013]; 35(i):[8i-9]. Disponible en: http://link.springer.com/article/10.1007/s10545-011-9415-4
17. Rodríguez M, Téllez N, CerbónMA, López M, Cervantes A. Metilación del ADN: un fenómeno epigenético de importancia médica. Rev. invest. clin, [online]. 2004 [citado 2013-09-18]; 56(1): [56-71]. Disponible en:
http://www.scielo.org.mx/scielo.php?pid=S0034-83762OO4OOOioooio&script=sci_arttext
18. Mendioroz M, Pulido L, Méndez I. Neuro-epigenética: metilación del ADN en la enfermedad de Alzheimer y otras demencias. Medicina Clínica [online] 2014 [acceso 18 de agosto de 2014] Disponible en: http://www.sciencedirect.com/science/article/PÜ/S0025775314002693 [ Links ]
19. Alien E, Horvart S, Tong F, Kraft P, Spiteri E, Riggs AD et al. High concentrations of long interspersed nuclear element sequence distin-guishmonoallelically expressed genes. PNAS [online] Agosto 19 2003 [acceso 26 de agosto de 2013]; 100(17):994O-5. Disponible en: http://www.pnas.org/content/100/17/9940.short
20. Murray R, Bender DA, Botham KM, Kenel-lyPJ, Rodwell VW, Weil PA. Harper bioquímica ilustrada. 29a Ed. Me Graw Hill; México DF: 2013. pp.257-60 [ Links ]