








Serviços Personalizados
Artigo
 Artigo em PDF
Artigo em PDF Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
Links relacionados
 Citado por SciELO
Citado por SciELO
 Similares em SciELO
Similares em SciELO
Bookmark
Revista SCientifica
versão impressa ISSN 1813-0054
SCIENTIFICA v.12 n.1 La Paz 2014
ARTÍCULO ORIGINAL
Transtornos de diferenciación sexual con genitales ambiguos en pacientes del instituto de genética La Paz - Bolivia. Periodo 2002 - 2012. Estudio Clínico - Genético
Disorders of sex development with ambiguous genitalia in patients of genetics institute of La Paz - Bolivia. Period 2002-2012 clinical - genetic study
Geraldine Montero Camacho1 Gonzalo Taboada 2 Erika La fuente 3 Beatriz Luna Barrón 4 Ana Rada 5
1 Estudiante de Medicina, Universidad Mayor de San Andrés
2 Master en Genética
3 Bioquímica Master en Genética
4 Especialista en Genética médica
5 Bioquímica farmacéutica.
Correspondencia a: Geraldine Montero Camacho
E-mail: gera_m@hotmail.es
Resumen
Introducción: La diferenciación sexual es un proceso complejo, dinámico y genéticamente controlado que culmina con la formación del fenotipo masculino o femenino. Los trastornos de diferenciación sexual (TDS) constituyen una condición congénita debida a defectos genéticos, cromosómicos, gonadales o fenotípicos que ocurren durante las etapas de la diferenciación sexual.
Objetivo: Investigar las características clínico - genéticas de presentación de TDS en pacientes del Instituto de Genética (IG) La Paz - Bolivia en el periodo 2002 -2012. Método y materiales:- Se revisaron las historias clínicas pertenecientes a pacientes con TDS y con genitales ambiguos. En total se revisaron 121 historias clínicas. Se descartaron 56 por falta de datos, diagnósticos no referentes al tema y falta de historia clínica. Resultados: Las características clínicas de ambigüedad genital más frecuentes fueron: micropene 5296, hipospadia 32,396, criptorquidia 30,896, hipertrofia de clítoris 18,596. Los TDS encontrados: TDS 46,XY 47,696, TDS,XX 4096, TDS por anomalías cromosómicas tipo síndrome de Turner 6,296, tipo síndrome de Klinefelter 1,596 y tipo quimerismo en un 3,196. La discordancia sexo asignado-cariotipo se dio en 30,796 de los casos: individuos con cariotipo 46,XX considerados masculinos, individuos 46,XY considerados femeninos, y casos de mosaicismo. Se encontró una frecuencia de TDS de 3,996 del total de consultas en el Instituto de Genética. Conclusión: Los TDS constituyen una condición congénita frecuente en nuestra población, que repercute en el aspecto físico, psicológico y social del paciente que lo padece. Por lo que es fundamental conocerlos y diagnosticarlos para brindar un óptimo tratamiento que será multidisciplinario.
Palabras clave:
Trastornos de diferenciación sexual, genitales ambiguos.
Abstract
Introduction: Sexual differentiation is controlled complex, dynamic and genetically process leading to the formation of the male or female phenotype. Disorders of sexual differentiation (TDS) are due to a congenital condition, due to genetic defects, chromosomal, or phenotypic gonadal occurring during different stages of sexual differentiation. Objective: To investígate the genetic and clinical presentation of TDS in patients of the Institute of Genetics (IG) La Paz - Bolivia in the period 2002 -2012. Method and materials: The medical records belonging to patients with TDS or with ambiguous genitalia were reviewed. A total of 121 medical records were reviewed. 56 were discarded due to lack of data, not related to the subject diagnosis and absence of medical history. Results: The most frequent clinical features of ambiguous genitalia were: micropenis 5296, 32.396 hypospadias, undecended testes 30.896,18.596 clitoral hypertrophy. Found TDS: TDS 46, XY 47.696, TDS, XX 4096. TDS whit chromosomal abnormalities: Turner syndrome 6.296, Klinefelter syndrome 1.596, and chimerism 3-196. The sex assigned-karyotype discordance occurred in 30.796 of cases: individuáis with karyotype 46, XX male considered, individuáis 46, XY female considered, and cases of mosaicism. TDS had a frequency of 3-996 of all medical consultation in the Genetics Institute. Conclusión: TDS are a common congenital condition in our population, affecting the physical, psychological and social aspects of the patient who has it. So it is essential to know and diagnose for optimal treatment shall be multidisciplinary.
Keywords:
Disorders of sex developmentl, ambiguous genitalia.
INTRODUCCIÓN
La diferenciación sexual es proceso complejo, dinámico y genéticamente controlado. Consecuencia de una serie de interacciones de señales genéticas, hormonales y celulares que culmina con la formación del fenotipo masculino o femenino. Este proceso puede verse afectado en cualquiera de sus tres etapas: genética, gonadal y fenotípica culminando en un TDS en el individuo1.
GENES IMPLICADOS EN LA DIFERENCIACIÓN SEXUAL
La diferenciación sexual inicia en el momento de la fecundación, definiendo el sexo cromosómico1. Las gónadas humanas son bipotenciales hasta las 6 primeras semanas de vida, es decir que pueden diferenciarse tanto a ovarios como a testículos. Esta diferenciación se da por un proceso molecular que implica varios genes:
Gen SRY (Sex- determining región Y).- se localiza en Yp11.3 y es considerado el gen determinante testicular. A los 41 días de gestación la expresión del gen SRY alcanza su máximo pico de expresión e inicia la diferenciación sexual. Produce un plegamiento del DNA permitiendo la interacción de regiones distantes que continuaran con la diferenciación testicular. Este gen se encontró translocado en individuos con fenotipo masculino y cariotipo 46,XX; así también se detectó su ausencia en individuos con fenotipo femenino y cariotipo 46,XY 2,3,4.
Gen SOX9 (SRY- Related HMG-Box Gene9) es el gen blanco del gen SRY. Se encuentra eni7q24.3-q25.i. Su expresión se ve incrementada después de la acción de SRY. SOX9 tiene un papel fundamental en el desarrollo y función de las células de Sertoli y por lo tanto para la expresión de HAM (Hormona antimuleriana). La delección se SOX9 causa reversión sexual masculina a femenina; su duplicación causa reversión sexual femenina a masculina. Las mutaciones heterocigotas de este gen causan Displasia Campomélica síndrome donde individuos 46,XY tienen fenotipo femenino además de alteraciones esqueléticas3,4.
GenDAXi Es un gen localizado en Xp2i.3. Necesita una dosis precisa para la correcta expresión del gen; ya que una duplicación de la región que contiene a DAXi (doble dosis de este gen), en varones XY interfiere con la formación del testículo y ocasiona reversión sexual sensible a dosis. DAXi ha sido identificado como antagonista de SRY, SOX9 y SFi en modelos murinos1,3,4.
Gen WTi (Wilms tumor 1) localizado en npi3, es un gen supresor tumoral, que cuando esta inactivado se relaciona con el Tumor de Wilms. Incrementa la estabilidad de SRY2. Sus mutaciones están relacionadas con diferentes síndromes: Síndrome WAGR: Tumor de Wilms, aniridia, malformaciones genitourinarias y retraso mental. Síndrome de Denys-Drash que puede presentarse con genitales ambiguos, o llegar al extremo de fenotipo femenino con cariotipo 46, XYi' 3,4.
Gen NR5A1/ SFi (factor esteroidogénico) Su locus es 9q33. En estudios murinos se comprobó que SFi se expresa primero para estimular a SRY y posteriormente para potenciar la expresión se SOX92.La deleción total de SF1 ocasiona la ausencia de gónadas y glándulas suprarrenales. Las mutaciones de SF1 causan insuficiencia suprarrenal primaria que se puede presentar con genitales ambiguos4.
Gen RSPO1(R-sponding family,memberi) Antes se creía que el desarrollo del ovario se daba por la ausencia o falta de expresión de genes que determinan el desarrollo testicular, en particular la ausencia de SRY. Actualmente se reconoce que el desarrollo del ovario en un proceso activo dirigido por genes que determinan su desarrollo y no así un proceso pasivo. El gen RSPOi está localizado en ip34,3 y ha sido propuesto como el gen determinante ovárico. RSPOi reprime la determinación testicular al degradar a SOX9. Las mutaciones que inactivan totalmente a este gen causan reversión sexual XX, hiperqueratosis palmoplantar y son pacientes son fenotípicamente masculinos4.
Gen WNT4 localizado en ip36. Sus funciones incluyen formación de la vasculatura sexo específica e incremento de la expresión de DAX12,4. La pérdida de función de ambas copias produce el síndrome SERKAL. Este último es incompatible con la vida y se caracteriza por genitales ambiguos, puede existir ovotestes, malformaciones cardiacas y pulmonares, agenesia renal e hipoplasia suprarrenal4.
Gen FOXL2 Ubicado en 3q23. Se piensa que es esencial para la determinación sexual femenina y el desarrollo ovárico temprano. Activa a CYP19 que codifica la enzima aromatasa que transforma los andrógenos en estrógenos. Las mutaciones ocasionan el síndrome de blefarofimosisptosis-epicantoinverso que puede asociarse a insuficiencia ovárica primaria4.
CARACTERÍSTICAS CLÍNICAS DE AMBIGÜEDAD GENITAL
Micropene.- Se define como un órgano morfológicamente y funcionalmente normal pero con una longitud por debajo de 2,5 Desviación Estándar de la media para su edad. En un recién nacido a término la longitud normal es de 3,5 +- 0,4 cm, estos valores antropométricos varían de acuerdo a raza y país5. Si el pene mide menos de 2,5 -2cm es considerado como micropene. En un adulto se considera micropene: longitud menor a 7cm en erección. Algunas causas de micropene son: hipogonadismo, deficiencia de 5 alfa reductasa, o insensibilidad parcial a los andrógenos5,6.
Hipospadias.- Defecto congénito en el que el meato uretral está localizado en cualquier sitio desde el glande hasta el periné a lo largo de la cara ventral del pene. En el sexo femenino este defecto corresponde a la aplasia de la cara posterior de la uretra. Diversos defectos genéticos, en especial los genes que codifican los receptores de andrógenos se asocian a la presencia de hipospadias5.
Epispadias.- En el varón el meato uretral se abre en la cara dorsal del pene. En el sexo femenino la epispadia consiste en la aplasia de la cara antero-superior de la uretra7.
Criptorquidia.- Es la ausencia de al menos uno de los testículos en el escroto. Lo más frecuente es que el testículo criptorquídico se localice a lo largo del trayecto habitual de descenso. La criptorquidia puede aparecer aisladamente, asociada a otras anomalías congénitas, o ser signo de endocrinopatías o de alteraciones cromosómicas (Síndrome de Klinefelter, Noonan)15.
Anorquia.- Testículo realmente ausente. No es posible encontrar la gónada incluso tras el empleo de las pruebas complementarias, y de la cirugía.
Atrofia testicular.- El desarrollo del testículo inicialmente es normal y la atrofia se debe a influencias externas. En el caso de los niños la atrofia testicular es consecuencia de: criptorquidia no corregida, síndrome de Klinefelter, síndrome de Noonan, hipogonadismo hipogonadotrófico8.
Hipogonadismo.- Situación clínico patológica que consistente en una hipofunción gonadal, ocasionada por un grupo heterogéneo de trastornos que interfieren en el normal funcionamiento del eje hipotalámico-hipofisario-gonadal. El hipogonadismo se diferencia en hipergonadotrópico (con fallo de función en la gónada) e hipogonadotrópico (con fallo en hipotálamo y/o hipófisis). Las causas son muy variadas y entre las causas genéticas se encuentran mutaciones del gen del receptor de LH o alteraciones del gen DAX i8. No existen datos clínicos de hipogonadismo en la infancia dado que el hipogonadismo se hace evidente solo ante la falta o retraso del desarrollo puberal9.
Hipoplasia de labios mayores y menores.- Se da un desarrollo incompleto de los labios mayores y menores, se presentan con aspecto marcadamente infantil. Se presenta en síndromes como: síndrome de Turner y Prader-Willi.
Fusión de labios.- La fusión de labios mayores se debe a exposición a andrógenos durante el desarrollo embrionario. Dependiendo del grado de exposición a andrógenos incluso pueden llegar a una fusión total y apariencia escrotal.
Hipertrofia de clítoris.- Es de las características de ambigüedad sexual más frecuente, es un signo de virilización por el exceso de andrógenos, y es causa frecuente de asignación errónea de sexo en el recién nacido. La primera causa es la hiperplasia suprarrenal congénita4.
MATERIALES Y MÉTODOS
Investigación de corte trasversal; se revisaron todas las historias clínicas de pacientes registrados en los libros del Instituto de Genética con TDS o con características clínicas de genitales ambiguos. En total se revisaron 121 historias clínicas, de las cuales se descartaron 56: 17 debido a datos insuficientes, y 36 que figuraban en los registros como TDS pero el diagnóstico en la historia clínica correspondía a otras patologías, no referentes al tema y 3 debido a falta de historia clínica.
Los resultados obtenidos se procesaron con el programa estadístico SPSS en el cual se registraron 65 casos; y se realizó un análisis que incluye la interpretación de frecuencias univariadas y bivariadas. Se aclara que en este análisis existen 2 casos registrados como "perdidos del sistema", debido a que no se contaba con todas las variables analizadas. Sin embargo estos casos fueron tomados en cuenta en el estudio debido a su relevancia como casos de adultos con discordancia sexo asignado-cariotipo.
RESULTADOS
El mayor número de casos se registró en la gestión 2012 con un porcentaje de 15,38%. En promedio se presentaron 6 casos de TDS por año. El 69,9 % de los pacientes son procedentes y residentes de la ciudad de La Paz, sin embargo se analizaron casos procedentes y residentes de todos los departamentos del país.
El 50% de los casos se presentaron con una edad entre o a 1 año. Casos con una edad de 2 a 4 años 12%, 5 a 10 años 5,8%, entre 11 y 15 años 9,2% y en un 10,8% casos entre 16 y 20 años.
La presencia de micropene como característica de ambigüedad genital fue la más frecuente en los casos con un porcentaje de 52%. La hipospadia en segundo lugar con 32,3%; la localización más reportada fue la peneoescrotal con un 9,2%, seguida de la balanoprepucial con un 3.1%. La criptorquidia fue la tercera característica de ambigüedad sexual en obtener mayor porcentaje con una presentación de 30,8%. La hipertrofia de clítoris se presentó en un 18,5%. (Figura 1)

Solo se reportaron 5 (7,7%) casos con hipogonadismo y de ellos solo uno contaba con los exámenes para clasificarlo como hipogonadotrófico. La fusión de labios solo se presentó solo en 3 (4,6%) casos.
En la revisión también se encontraron características clínicas de ambigüedad sexual que no fueron tomadas en cuenta en el inicio de la investigación. Estas tuvieron una presentación de 3.1% cada una y se citan a continuación:
Escroto en chai, Escroto liso sin pliegues, Ginecomastia, Labios mayores de apariencia escrotal, Pseudovagina, Ausencia de vello genital, Vagina pequeña.
Se reportaron cariotipos correspondientes a: cariotipos normales con un 38,5% para 46,XX y un 43,1% para 46,XY; síndrome de Turner 45>X (3,1%), y por mosaicismo: 46,XY/45,X (3,1); El síndrome de Klinefelter se presentó en mosaicismo: 46,XX/47,XXY (1,5%). El mosaicismo 46,XX/46XY se presentó en un (3,1%). Con un 1,5% cada uno se encontraron diferentes cariotipos. (Figura 2)

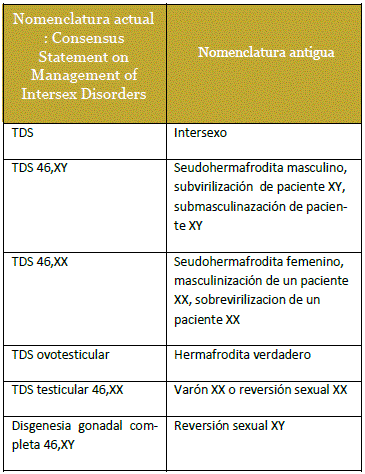
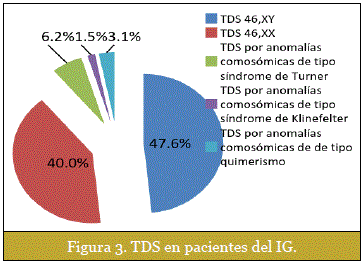
El estudio tomó en cuenta la clasificación del: Consensus Statement on Management of Intersex Disorders llevado a cabo en Chicago el año 200510 (Tabla 1) y sus modificaciones realizadas el año 201011. Deacuerdo a esta clasificación el TDS más frecuente fue: TDS 46,XY 47,6%. (Figura 3).
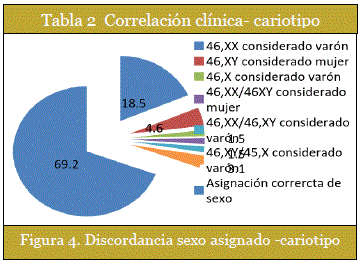
Se rescataron 20 casos de discordancia sexo asignado-cariotipo, (sexo de ingreso - cariotipo) que representan el 30,7% de los casos. Son casos de asignación errónea de sexo al individuo por los familiares debido a la ambigüedad genital. (Figura 4)
Correlación clínica- cariotipo
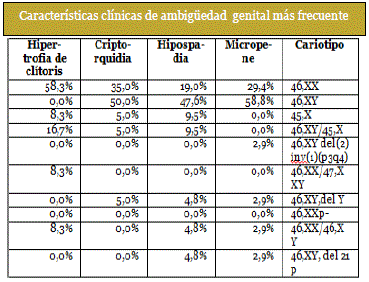
Este análisis se realizó para poder relacionar las características clínicas de ambigüedad genital con mayor frecuencia en el estudio con un cariotipo determinado:
Micropene.- Para un cariotipo 46,XY se encontró 58,8% de presencia de micropene. Llama la atención que el segundo porcentaje más elevado: 29,4 % corresponda a valoraciones de micropene con cariotipo 46,XX.
Criptorquidia.-se presentó en un 50% para cariotipo 46,XY; 35% en pacientes con cariotipo46,XX. Un 5% de las criptorquidias fueron valoradas en síndrome de Turner.
Hipospadia.- se presentó en un 47,65% para cariotipo 46,XY, y en un 19% para cariotipo 46,XX.
Hipertrofia de clítoris.- La hipertrofia de clítoris se presentó en 58,3% en individuos con cariotipo 46,XX. Un 16,7% corresponde a pacientes con cariotipo 46,XY/45,X. (Tabla 2)
DISCUSIÓN
El 50% de los casos se presentaron con una edad entre o a 1 año, edad crítica en la que los TDS constituyen una emergencia al momento de la asignación correcta de sexo al individuo. Los casos con una edad entre 11 y 15 se detectan generalmente por hipogonadismo. Los casos entre 16 y 20 años etapa en la cual el desarrollo de caracteres sexuales secundarios debería completarse y la identidad sexual del individuo está definida conllevan mayores dificultades en el manejo psicológico del paciente12.
Si bien la hipertrofia de clítoris se presentó en un 18,5% se debe aclarar que se encontraron casos con descripción de micropene con un cariotipo posterior 46,XX, considerando esto se elevaría el porcentaje de la presentación de hipertrofia de clítoris. Sin embargo para el estudio se consideró que los datos debían registrarse y analizarse así como se hallaban en las historia clínicas.
Los cariotipos normales obtuvieron los porcentajes más altos con un 38,5% para 46,XX y un 43,1% para 46,XY, lo que revela la necesidad de aplicar métodos citogenéticos moleculares para identificar alteraciones génicas que no son evidentes ante los métodos convencionales de la citogenética clásica.
Un 36,9% de los pacientes llegaron al Instituto de Genética sin un diagnóstico presuntivo, este valor elevado remarca la necesidad del personal de salud de conocer más sobre los TDS. Un 18,5% de casos corresponde a individuos con cariotipo 46, XX que fueron considerados masculinos, dato que revela la importancia del diagnóstico de la hipertrofia de clítoris y su principal causa: Hiperplasia suprarrenal congénita. En este sentido el análisis de correlación clínica-cariotipo es útil para esperar un cariotipo determinado de acuerdo a la característica clínica de ambigüedad genital que se encuentra en el paciente.
Idealmente y según recomendaciones internacionales el abordaje de un caso de TDS debe ser multidisciplinario con un equipo que debería incluir: pediatras con subespecialidad en neonatología, endocrinología, cirugía, urología, ginecología; además un médico genetista, apoyo psicológico y psiquiátrico, y un equipo que vele
la ética de las acciones a tomarse10. Evidentemente no es la realidad que se da en nuestros centros de salud y una vez más remarca la importancia del conocimiento de este tema para tratar adecuadamente a los pacientes que lo padezcan.
CONCLUSIONES
El 50% de los casos se dieron entre 0 -1 años, dato que no es extraño pues la asignación correcta del individuo es una emergencia y es en esta etapa en la que también se recomienda la realización de las cirugías necesarias al paciente. El trastorno de diferenciación sexual que se presentó con mayor frecuencia fue: TDS 46,XY en un 47,6%. En cuanto a la clasificación, es importante el uso de la nueva nomenclatura que evita términos antiguos que son confusos y hasta insensibles con el paciente y la familia.
Las características de ambigüedad sexual más frecuentes encontradas fueron: micropene 52%, hipospadia 32,3%, criptorquidia 30,8% y la hipertrofia de clítoris 18,5%. Características que deben ser identificadas y reportadas al momento del nacimiento.
La discordancia sexo de ingreso- cariotipo se dio en un 30,7% de los casos. Es sin duda un elevado porcentaje y constituye un problema serio tanto para el paciente como para la familia en varios aspectos. Algunos con mayor magnitud a determinada edad, como la gran responsabilidad de los padres para elegir el sexo correcto del RN. O ya en la edad adulta del paciente con una identidad sexual bien definida.
Se calculó que del 100% de las consultas realizadas en el Instituto de Genética 3,9% corresponden a TDS lo que en una mínima muestra revela que los TDS constituyen una condición congénita frecuente en nuestra población, que repercuten en el aspecto físico, psicológico y social del paciente que lo padece.
Se espera que este pequeño estudio incentive investigaciones de mayor envergadura sobre el tema de forma que logremos conocer y diagnosticar los TDS para brindar un óptimo tratamiento a los pacientes.
AGRADECIMIENTO
Infinitas gracias al Instituto de Genética y a los tutores que guiaron esta investigación por el apoyo y orientación incondicional.
REFERENCIAS
1. Larson A, Nokoff N, Travers S. Disorders of Sex Development: Clinically Relevant Genes Involved in Gonadal Differentiation. Discov Med 14(78):301-309. [ Links ]
2. McClelland K, Bowles J, Koopman P. Male sex determination: insights into molecular mechanisms. Asian Journal of Andrology 2012; 14 (1): 164-171 [ Links ]
3. Gónül Ógal.Current Concepts in Disorders of Sexual Development. J Clin Res Ped Endo 2011; 3(3):105-114 [ Links ]
4. Kofman S, Nieto K, Gómez L, Cervantes A. Aspectos genéticos, moleculares y celulares de la diferenciación gonadal. En: Del CastilloV, Rafael D, Uranga H. Genética clínica. México: El manual moderno; 1012.p. 269-279. [ Links ]
5. Perea L, Martinez- Aedo J, Nieto J, Prieto J. Intersexo, Hipospadias, micropene, criptorqui-dia. Sociedad española de endocrinología pediátrica 2008; 15(1) 3-22. [ Links ]
6. Guzmán J. Micropene. Rev Mex Urol 2008; 68(1):1-2 [ Links ]
7. Duque S, Rodríguez C, Páez J. Complejo ex-trofia-epispadias: Experiencia del instituto Infantil Roosevelt y en la clínica Shaio de Bogotá Colombia, urol.colomb. 2010; 19(3):111-117. [ Links ]
8. P. Martul. Hipogonadismo: diagnóstico y tratamiento. An Esp Pediatr 2000; 52 (1): 59-62. [ Links ]
9. Cañete Estrada R, Mata Rodríguez C, Aguilar Quintero M. Retraso puberal. Hipogonadismos. Protoc diagn ter pediatr.2011; 1 (1):2O5-17.
10. Peter A. Lee, Christopher P. Houk, S. Faisal Ahmed and Ieuan A. Hughes. Consensus Statement on Management of Intersex Disorders. Pediatrics 2006; 118 (2): 752-757. [ Links ]
11. Kofman S, Mayorga J, Venegas C, Lira S. Desórdenes del desarrollo sexual. En: Del CastilloV, Rafael D, Uranga H. Genética clínica. México: El manual moderno; 1012.p. 281-292. [ Links ]
12. Sandberg D, Gardner M, Cohen-Kettenis P. Psychological Aspects of the Treatment of Pa-tients with Disorders of Sex Development. Semin Reprod Med. 2012; 30(5): 443-452. [ Links ]