








Servicios Personalizados
Articulo
 Articulo en PDF
Articulo en PDF Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
Links relacionados
 Citado por SciELO
Citado por SciELO
 Similares en SciELO
Similares en SciELO
Bookmark
Archivos Bolivianos de Medicina
versión impresa ISSN 0004-0525
Arch.Boliv.Med. v.19 n.87 Sucre 2013
XERODERMA PIGMENTOSO
Autores:
Dra. Ramallo Jadue Fabiola (1); Dr. Ramallo Jadue Jorge (2).
(1) Dermatóloga - Inmunoalergóloga, Docente titular de Dermatología U.RPS.FX.CH.
(2) Médico Cirujano, Magister en Microbiología.
Recepción: 21/marzo/2012 Aceptación: 14/agosto/2012
RESUMEN
El xeroderma pigmentoso, constituye una patología derivada de una sensibilidad muy aumentada a la luz solar, con presencia de alteraciones en la pigmentación cutánea, piel seca, queratosis, telangiestasias. Un 20% de los pacientes presentan alteraciones neurológicas y oculares asociadas a la fotofobia, presentando una elevada predisposición al desarrollo de múltiples canceres de piel, (carcinomas espinocelulares, basocelulares), en una relación de 1000 veces mayor en relación a la población general. La etiología de la enfermedad es genética de carácter autosómico recesivo.
Se describe dos infrecuentes patologías genéticas, el síndrome de Cockaine y la Tricotiodistrofia similares al xenoderma pigmentoso. Por ello el estudio de las alteraciones genéticas presentes en estas tres entidades constituyen un modelo de gran utilidad para la comprensión de la importancia del daño y reparación del ONA en la génesis de los tumores de piel.
PALABRAS CLAVES:
Xenoderma pigmentoso, síndrome de Cockaine, Tricotiodistrofia, cáncer de piel.
SUMMARY
The xeroderma pigmentosum, is a disease resulting from a sensitivity greatly increased to sunlight, with the presence of alterations in skin pigmentation, dry skin, keratosis, telangiestasias.
20% ofpatients present with neurological and related eye photophobia, presenting a high predisposition to develop multiple skin cancers (squamous cell carcinoma, basal cell), at a ratio of 1000 times higher compared to the general population.
The etiology ofthe disease is genetic in nature autosomal recessive. It describes two rare genetic disease, syndrome
Cockaine and Trichothiodystrophy similar to xenoderma pigmentosum. Therefore, the study of genetic alterations found in these three entities are a very useful model for understanding the significance of the damage and repair of DNA in the genesis of skin tumors.
KEYWORDS:
Xenoderma pigmentosum, Cockaine syndrome, Trichothiodystrophy, skin cancer.
INTRODUCCIÓN.-
El xeroderma pigmentoso es un modelo útil para el estudio de las alteraciones moleculares que impiden la correcta reparación del ONA dañado por los RUV.
Los pacientes afectados por xeroderma pigmentoso presentan un riesgo de padecer tumores dermatológicos mil veces mayor que la población general
El xeroderma pigmentoso (XP) es una enfermedad de muy baja prevalencia. Se considera que aparece en un caso cada 250.000 personas. La enfermedad deriva de una sensibilidad muy aumentada a la luz solar, clínicamente se caracteriza por la presencia de alteraciones en la pigmentación cutánea (hipopigmentacion e hiperpigmentacion), con producción de lesiones dermatológicas como piel seca, queratosis actínica y telangiectasias. En un 20% a 30% de los pacientes se observan modificaciones neurológicas progresivas y oculares asociadas a fotofobia. Simultáneamente se presenta una elevada predisposición al desarrollo de múltiples canceres de piel. Los de aparición más frecuente son los carcinomas espinocelulares y basocelulares en las zonas expuestas a la luz solar y los melanomas de distribución más variada, aunque también se observan tumores de la cámara anterior del ojo y en la punta de la lengua. Se estima que el riesgo de padecer tumores dérmicos en los pacientes afectados por XP es 1000 veces mayor que en la población general. El diagnostico de la enfermedad es confirmado por la investigación del ONA en cultivos de fibroblastos, y los fundamentos del tratamiento se basan en la protección de los pacientes de los efectos de la radiación ultravioleta. La etiología de la enfermedad es genética, siendo heredada con carácter autosómico recesivo. La fisiopatología depende de I a incapacidad de los pacientes afectados de reparar las lesiones de ONA generadas por la radiación ultravioleta. El 80% de los pacientes presentan un defecto en la vía de reparación por escisión de nucleótidos (via NER). El 20% restante puede reparar su material genético eficientemente pero la replicación genética se halla interrumpida en los sitios mutados por la radiación ultravioleta. Al primer grupo se lo denomina como portador de XP clásico y al segundo de variantes de XP.
Se han descripto otras dos infrecuentes enfermedades genéticas en las cuales se han hallado defectos de la vía NER con fotosensibilidad. La primera de ellas es el síndrome de Cockaine y la segunda es una forma fotosensible de tricotiodistrofia(TTO). En el primero de estos cuadros, los pacientes se caracterizan por presentar retardo del crecimiento y enanismo, alteraciones de los órganos de los sentidos (degeneración retiniana y sordera), y desmielinización que produce una degeneración neurológica progresiva por lo que fallecen a temprana edad con apariencia de envejecimiento. En la segunda afección los enfermos también son de corta estatura con apariencia y con retardo mental, a lo cual se suma la aparición de icitioso y debilidad de cabellos y uñas e infertilidad. Por último, se han hallado algunos casos de individuos que presentaban características fenotípicas de XP asociados a las del Síndrome de Cockaine.
Una de las facetas más llamativas de estas dos afecciones mencionadas es que, a pesar de que su etiología está relacionada con defectos en la vía NER, no se observa en las personas afectadas un incremento en el riesgo de tumores dermatológicos. Por otro lado, en el XP se hallan alteraciones del sistema nervioso central que no dependen de la fotosensibilidad, lo cual sugiere la existencia de otros mecanismos fisiopatológicos diferentes de la dificultad en reparar el ONA.
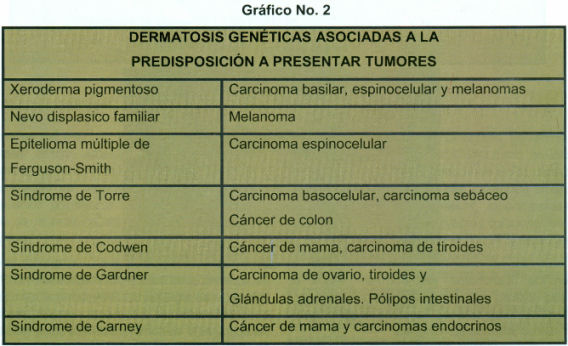
Se acepta que en los individuos de raza blanca el desarrollo de canceres de piel se halla relacionado con mecanismos fisiopatológicos similares derivados de la sobrexposición a la luz solar. Por ello, el estudio de las alteraciones genéticas presentes en el Xp, en el síndrome de Cockaine y en la tricotiodistrofia constituye un modelo sumamente útil para la comprensión de la importancia del daño y la reparación del ONA en la génesis de los tumores.
Asimismo, resulta de gran importancia resolver cual es el motivo por el cual el síndrome con similares deficiencia genéticas producen presentaciones clínicas tan disimiles entre si.
DESCRIPCIÓN.
RUVygenoma. El espectro de la radiación ultravioleta puede ser dividido según su longitud de onda en radiación UVA, UV-B y UV-C. La radiación UV-C tiene una longitud de 100 a 280 nm. El ozono atmosférico constituye una barrera de particular importancia que impide el paso de esta radiación, por ello se considera que su contribución al desarrollo de cáncer de piel es despreciable excepto por factores de exposición ocupacional. La radiación UV-A es la que alcanza la superficie terrestre en mayor proporción, estimándose en un 95% de la radiación total (aunque varía según la latitud) y su longitud de onda oscila entre los 315 y 400 nm. El efecto de sus rayos sobre el material genómico es básicamente de orden oxidativo, como la formación de timidinglicol y 8-oxodesoguanina. Por ello se ha calculado que su contribución al riesgo de tumores dermatológicos es baja. Por último la radiación UV-B de longitud de onda de 280 a 315 nm constituye solamente alrededor del 5% de la que alcanza la superficie terrestre. Sin embargo, su efecto sobre el ONA es sumamente perjudicial, aceptándose que es la radiación que más incrementa el riesgo de canceres en la piel.

Referencia. La reparación del ONA se produce mediante dos vías, la TCR (reparación de transcripción apareada) y la GGR (reparación global del genoma). En esta última, el reconocimiento de la zona afectada se produce por el complejo XPC asociado a HHR 23B Y, en el caso de la reparación TCR, el reconocimiento se produce por la RNAPII asociada al complejo CSA-CSB 2) Por una u otra vía, el complejo que reconoce al ONA alterado se une a otro conformado por TFIIH-XPA-RPA, que produce el desenrollamiento de la doble hélice. Esto permite la incisión del ONA por parte de las endonuc/easas XPFyXPG 3) Una vez retirado el ONA alterado se resintetiza el polidesoxinucleotido por complementariedad. 4) Síntesis del nuevo ONA
Las lesiones genómicas producidas por estos rayos se dividen en dos grupos. Las más común (75% de los daños) es la formación de dímeros de pirimidina-ciclobutano (COP), y la de menor frecuencia (25% restante) es la generación de distintos fotoproductos de 6-pirimidina-4-pirimidona ((6-4)PP). Se puede observar la ligadura de dos timinas (5'C-C) o de una citosina con una ti mina (5'C-T, 5'T-C). La unión de dos timinas es la lesión más frecuente de las halladas en humanos y aparentemente no constituye una alteración de gran importancia mutagenica. En cambio, los dímeros de citosina o de citosina-timina son altamente mutagenicos. La doble transición citosina-citosina a timina-timina (C-C T-T) está considerada como una mutación específicamente derivada de la acción solar, por lo que se le ha denominado la huella digital de la lesión por radiación ultravioleta.
La reparación de los fotoproductos es sumamente eficiente en los seres humanos, a diferencia de CPO que es mucho más lenta y dificultosa.
Respuesta celular al daño del ONA
En respuesta a la alteración genómica inducida por las radiaciones UV o por otros factores (hipoxia, radiaciones ionizantes) se desencadena una serie de procesos protectores como supresión inmunitaria transitoria, acumulación de proteína p53 y sobreexpresion de genes que guían a la célula a la apoptosis, y mecanismo de reparación del ONA por la vía NER

La función del gen p53 es crucial en la respuesta defensiva celular. Ante la aparición de un daño genómico se incrementa en forma notable la concentración de proteína p53 a través de dos mecanismos posteriores a la transcripción: la vía media de la proteína aumenta y la traducción se multiplica.
Una vez incrementados sus niveles, la proteína se une a diversos genes como Gadd45, el oncogen mdm2, el gen que origina a la proteína bax y al p21, y estimula su transcripción. El producto del p21 se une al complejo ciclina/ccdk e inhibe la actividad de cdk que es necesaria para el pasaje de la célula de estado G1 a S.A través de estos mecanismos el p53 genera la detención del ciclo celular, sin embargo no es la única forma de actuar del gen p53. También se ha demostrado que conduce a la célula hacia la apoptosis (muerte celular programada). Para ello se activan una serie de proteasas denominadas caspasas de la familia ICElCed-3. Estas proteasas tienen afinidad por los residuos de ácido aspártico de los sustrato y c1ivan ciertas moléculas proteicas especifica como proteincinasas, topoisomerasa 1, AOP-ribosa polimerasa y diversos componentes del citoesqueleto. No se conoce con total certeza cuales son las enzimas cuya destrucción es el punto crítico para lograr la muerte celular, pero si está claro que la acción de las proteasas conduce a este resultado. Por medio de estos dos mecanismos-detención en fase G1 Y apoptosis-el gen p53 logra inhibir la multiplicación celular cuando existen modificaciones anormales de AON. Por lo tanto, no resulta casual que su inactivación sea necesaria para el crecimiento tumoral, y que sea el gen que se encuentra más frecuentemente mutado en la patología oncológica humana.

Referencia. Epitelioma basocelular, proliferación de células basaloides que se disponen en nidos rodeados por empalizada periférica

Referencia: Carcinoma espinocelular.- Nidos de células atípicas con marca acidofilia citoplasmática esbozando perlas corneas, inmersas en tejido fibroso con intenso infiltrado mononuclear
En cuanto a la reparación del ONA a través de la vía NER se observado que esta vía funciona bajo dos formas cinéticas diferentes, según la función del ONA dañado. Si la zona a reparar es de aquellas que rápidamente transcribe la RNA polimerasa 11, su reparación es más rápida, más completa y más eficiente a través de un, mecanismo denominado de reparación de transcripción apareada (TCR). En cambio, si las regiones del ONA dañadas son distintas a las señaladas, se activa otra vía NER alternativa conocida bajo el nombre de reparación global del genoma (GGR)
La primera etapa necesaria para iniciar la modificación de ONA mutado es el reconocimiento de la existencia de un sitio lesionado. En la vía GGR este reconocimiento se produce a partir de la unión de las proteínas XPC al sitio de la alteración. Esta proteína se halla formando un complejo con la proteina HHR23B, y al localizar y unirse al DNA generan un cambio conformacional de la molécula. Luego, la interacción de este complejo con el factor de iniciación de transcripción IIH (TFIIH) Y la proteína XPA asociada a proteína de replicación A (RPA) generan el desenrollamiento de la doble hélice en la región adyacente a la alteración. En la vía TCR la función de la proteína XPC es cumplida por otra molécula, la proteína RNAPII cuya conformación podría ser modificada por los complejos proteicos CSA y CSB. A partir de esta etapa ambas vías iniciales de reparación GGRy TCR confluyen en una única ruta final. La incisión del DNA es realizada a una distancia de cinco a seis nucleótidos por debajo de la mutación por una endonucleasas, la XPG, y en una unión distante 20 a 22 nucleótidos por encima del daño por otra endonucleasas, el complejo XPF, ERCC1.
LA CÉLULA SE DEFIENDE DEL DAÑO GENÓMICO POR MEDIO DEL INCREMENTO DE LA PROTEíNA p53 Y DE LAS VíAS DE REPARACIÓN DEL ONA.
Se considera que un espaciamiento tan preciso es logrado por la interacción de todas las moléculas involucradas que forman un verdadero soma de reparación conformado por la ligadura de la XPA con la ERCC1 y la XPG con la TFIIH y el CSB. Luego la cadena dañada es liberada y diversas proteínas actúan en la replicación para la síntesis de nuevo DNA. El espacio es rellenado por acción de la DNA polimerasa delta y/o épsilon y el extremo S'es unido por la DNA ligasas ,l
Se ha encontrado que los pacientes afectados por XP clásico presentan alteraciones en siete grupos de genes de complementación (XPA, XPB, XPC, XPD, XPE, XPF Y XPG) mientras que los afectados por variantes de XP se halla alteración del XPV Dentro del pequeño grupo de pacientes que ha podido ser clasificado, se observo que las alteraciones mas frecuentes ocurren a nivel del XPA, XPC y XPV y en menor medida en el XPD. En el síndrome de Cockaine se describe la alteración del CSA y del CSB, siendo esta ultima la de mayor ocurrencia relativa.
La apariencia fenotípica de TTD es producida por la modificación del XPB y del XPD. En los últimos años se ha descubierto que estas dos proteínas forman parte del complejo TFIIH. Este complejo que cumple funciones tanto en la vía NER como en la transcripción esta integrado por nueve subunidades, dos de las cuales son las XP señaladas. Ambas cumplen función de helicasas, la XPB en sentido 3' 5' Y la XPD en sentido inverso 5' 3'. El heco de que estas proteinas participen de ambos procesos-de reparacion y de transcripcion- justificaria las diversas presentaciones clinicas de los pacientes con deficiencia de ellas. En un estudio reciente se demostro que los pacientes con mayor severidad de sintomas por deficiencia de XPB eran aquellos cuyo TFIIH resultaba mas defectuoso en su funcion en la transcripcion. Asimismo se hallo que en relacion con el XPD, los pacientes con fenotipos mas severos eran los tenian mutacion en el extremo 3 del gen.
Aunque los procesos descriptos son de suma importancia en el desarrollo de la patologia oncologica dermica, no se debe descartar la existencia de otros mecanismos involucrados ya que el crecimiento tumoral es multifactorial. La activacion de oncogenes, la falla en el funcionamiento de genes supresores como elp53yla supresion de la respuesta inmunitaria de celulas NK y de la produccion de interferon del huesped ayudan a entender mejor la susceptibilidad individual a la radiacion UV
REFERENCIAS BIBLIOGRÁFICAS
1. HuJC.; SadeghiP; Pinter-Brown L. y col.: Cutaneous side effectsof epidermal growth factor receptor inhibitors:Clinical presentation,pathogenesis, and management. JAm. Acad Dermatol 2007;Vol 52:121. [ Links ]
2. SegaertS.; Cutsen V: Clinicalsigns,pathophysiology and management of skin toxicity during therapy with epidermal growth factor receptor inhibitors. Annals of Oncology 2005;16:1425-1433. [ Links ]
3. Marini M.; Noriega G.; Galimberti D.; Marini M,secundaria a erlotinib. Act Terap Dermatol 2007;30:20. [ Links ]
4. Cowen E.: Epidermal growth factor receptor inhibitors:A new eraof drug reactions in a new era of cancer therapy. J Am.Acad. a silver Lining? J Clin Oncol 2005: 23:5235-5246. [ Links ]
7. Guhl G.; Gonzalez A.; Dauden E.: Efectos cuténeos de los inhibidores del receptor del factor de crecimiento epidérmico. ActasDermatosifiliogr 2006;97(5):296-310. [ Links ]
8. DeWitt C.; SiroyA.; Stone S.: Acneiform eruptions associated withepidermal growth factor receptor-targeted chemotherapy.J Am AcadDermatol 2007;56: 500-5. [ Links ]