








Servicios Personalizados
Articulo
 Articulo en PDF
Articulo en PDF Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
Links relacionados
 Citado por SciELO
Citado por SciELO
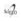 Similares en SciELO
Similares en SciELO
Bookmark
Archivos Bolivianos de Medicina
versión impresa ISSN 0004-0525
Arch.Boliv.Med. v.14 n.82 Sucre 2010
El Rol del lodo en el Crecimiento y Regulación Tiroidea
Thomasa, Lisa1; Pisarev, Mario1 y Juvenal, Guillermo1
' División Bioquímica Nuclear; Departamento de Radiobiología, Comisión Nacional de Energía Atómica, Argentina
El iodo juega un importante papel en la bioquímica y fisiología tiroidea. Es indispensable para una adecuada biosíntesis hormonal, ya que es el único factor limitante de la misma en condiciones normales.
El iodo es ingerido con los alimentos (carne, leche, complejos vitamínicos, alimentos marinos, sal de mesa) (Rousset & Dunn, 2004) y es absorbido en el intestino delgado. La mayoría del iodo molecular es reducido a ioduro luego de su absorción intestinal. El anión se encuentra normalmente en el plasma en una concentración de 10 ug/l, y en pequeñas cantidades en huesos y glóbulos rojos (Wynn, 1961). El riñón es el encargado de eliminar el exceso de iodo, aunque puede reabsorberlo. Esta reabsorción aumenta en los casos de hipotiroidismo y disminuye en el caso opuesto (Wynn, 1961). Las glándulas salivares también pueden captar iodo, así mismo lo hacen las glándulas mamarias y otros órganos (Rousset & Dunn, 2004). Si bien la TSH es el principal regulador de la función y crecimiento de la glándula tiroides (Dumont et al., 1992), el contenido intratiroideo de iodo es un importante regulador de estos parámetros.
Las acciones del iodo sobre el crecimiento tiroideo han sido uno de los efectos más estudiados. La depleción de iodo genera hipersensibilidad a la acción de TSH. Bray describió en 1968 que en ratas hipofisectomizadas sometidas a una dieta pobre en iodo que el crecimiento glandular estimulado por TSH exógena es mayor respecto a animales que han recibido una dieta normal. Más aún cuando el contenido intratiroideo de iodo se encuentra significativamente disminuido se puede observar que los animales desarrollan bocio aún con niveles normales de TSH, lo que explica la coexistencia de bocio con niveles normales de TSH en zonas de la Argentina donde antaño existía endemia bociosa (Pisarev et al., 1970). La carencia de iodo, en diferentes regiones del mundo, causa un aumento de la incidencia de bocio en la población afectada, que se acompaña de un alteraciones en el recién nacido, alteración cerebral y osteomuscular, aumento de la frecuencia de infertilidad, abortos y de tumores tiroideos (Delange & Dunn, 2005).
Como mencionamos anteriormente la falta de iodo, trae como consecuencia un agrandamiento de la glándula (bocio), una mayor vascularización y una mayor sensibilidad a la TSH, y si estos mecanismos de compensación no alcanzan, se resiente la producción hormonal (Pisarev y Gartner, 2000, Panneels et al, 2009).
Por otra parte, el exceso de iodo tiene efectos opuestos. Se ha demostrado que el iodo es capaz de inhibir una larga lista de parámetros tiroideos, tanto en lo que respecta a la función como al crecimiento glandular. A este proceso intraglandular que modula la función tiroidea y la respuesta glandular a factores exógenos se lo ha denominado autorregulación tiroidea.
La autorregulación tiroidea resulta de mantener la secreción hormonal constante, aún frente a variaciones en la ingesta de iodo.
A medida que el aporte de iodo aumenta, se incrementa la biosíntesis de las hormonas tiroideas. Sin embargo, cuando la concentración de iodo supera un cierto límite se pierde la proporcionalidad entre ambos parámetros y se inhibe progresivamente la biosíntesis hormonal. A esta acción del exceso de iodo se la denomina efecto Wolf-Chaikoff (Wolf J et al., 1949).
Si bien en un comienzo el exceso de iodo inhibe varios parámetros tiroideos, esta acción desaparece si se continúa con la administración de iodo, fenómeno conocido como escape. Este fenómeno se explica porque el exceso de iodo inhibe su propia captación, provocando que al cabo de un tiempo la concentración de iodo intratiroidea disminuya, revirtiendo así la acción inhibitoria.
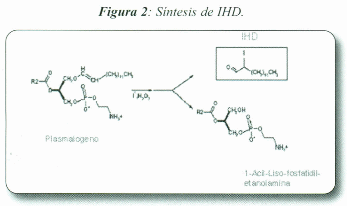
Es conocido que la captación de iodo se encuentra modulada fundamentalmente por la TSH. Sin embargo el iodo mismo constituye otro importante regulador. El exceso de iodo revierte la acción ejercida por la TSH tanto in vivo como in vitro (Pisarev & Gartner, 2000, Panneels et al, 2009). Por ejemplo, los efectos del iodo sobre la reacción de organi-ficación y acoplamiento de iodotironinas dependen de la duración y de la concentración de iodo. Wolf y Chaikoff observaron que cuando la concentración de iodo circulante alcanza un nivel crítico, en condiciones agudas existe un punto de inflexión en el incremento de la organificación, y el contenido de iodo organificado decrece rápida y progresivamente en función de la dosis de iodo ofertada. El bloqueo de la organificación es producido por la inhibición de los sistemas generadores de H202 (DUOX) y DUOX2) con la subsiguiente disminución de los niveles de H202. El bloqueo provocado por el iodo es temporal (fenómeno de escape), y luego la reacción alcanza los niveles normales a pesar de mantenerse elevada la oferta de iodo circulante.
El exceso de iodo inhibe su propia captación, porque se produce una disminución en la expresión del transportador de iodo (NIS). La inhibición en la expresión de NIS podría ser la responsable del fenómeno de escape del efecto Wolff-Chaikoff(Engetal., 1999).
A altas dosis de ioduro se inhibe la secreción hormonal (De Groot et al., 1956) y se produce también una disminución del flujo sanguíneo (Rognoni et al., 1984). La TSH regula el crecimiento y la función tiroidea a través de las cascadas de AMPc y la vía de Ca2+/PIP2 y el iodo intracelular regula estas dos vías (Laurent et al., 1989). El iodo inhibe la acumulación de AMPc en respuesta a TSH o forscolina, esta disminución provoca una inhibición de todos los efectos mediados por AMPc. Si el efecto es a largo plazo, desensibi-liza la célula al estimulo de TSH (Van Sande et al., 1975). In vitro se ha observado que además se inhiben otras variables tales como la actividad de la adenilato ciclasa (Cochaux et al., 1987, Van Sande et al., 1975), la síntesis de Tg (Pregliasco et al., 1996). la proteólisis de Tg (Pisarev, 1985), la síntesis de TPO y NIS, pero no del rTSH.
Además están inhibidos los procesos de pinocitosis (desde el lumen al citoplasma), la captación de glucosa y aminoácidos, la síntesis de ADN y ARN Y a altas dosis puede producir muerte celular por apoptosis.
El ioduro inhibe la cascada del AMPc y la acción del AMPc per se y la vía del Ca++/PIP2 a nivel de Gq o de la fosfolipasa C.
El exceso de iodo inhibe la proliferación celular, se ha propuesto que el iodo actuaría sobre el ciclo celular a nivel de pasaje de G¿ a G1 (Smerdely et al., 1995).
In vivo disminuye el flujo sanguíneo, y el crecimiento glandular (Pisarev, 1985). También están inhibidas las acciones autócrinas o parácrinas como las relacionadas con EGF, IGF-1 Y FGF.
Mecanismo de acción del iodo.
Se ha demostrado que para que el iodo ejerza su acción auto-rregulatoria debe ser incorporado a moléculas orgánicas. La mayoría de los parámetros tiroideos que son inhibidos por el exceso de iodo, son liberados de esta inhibición si se incuba in vitro o administra in vivo, simultáneamente con el exceso de iodo, drogas que bloquean la acción de la peroxidasa tiroidea, como el metilmercaptoimidazol (MMI) o el propiltioura-cilo (PTU). Se han postulado como intermediarios, iodopépti-dos, las mismas hormonas tiroideas (Juvenal et al., 1981) y lípidos iodados. De estos últimos se han identificado y caracterizado dos: la 6-iodo-l l,14-eicosatrienoico -o-Iactona (IL-d) (Boeynaems & Hubbard, 1980; Dugrillon, et al., 1990) y el 2-iodo-hexadecanal (1HD) (Panneels et al., 1996; Pereira et al., 1990).
Estudios in vitro e in vivo han demostrado que los iodolípidos reproducen los efectos de exceso de iodo, y esto es válido para el IHD (Panneels et al., 1994, Ohayon et al., 1994) y para la IL-o (Chazenbalk et al., 1988; Krawiec, et al., 1988). El ácido araquidónico es el precursor de la IL-o (Pisarev et al., 1985) y su síntesis se incrementa con el agregado de ácido araquidónico (Boeynaems & Hubbard, 1980); esta hipótesis
se confirmó en folículos porcinos, tiroides de rata y tejido humano (Dugrillon et al., 1994). lodolactona (IL-o):
En 1981 Boeynaems et al. demostraron que la lactoperoxida-sa era capaz de catalizar la iodinación de algunos lípidos poliinsaturados, resultando en la formación de iodolactonas (Boeynaems et al., 1981a,bc). El principal producto de la reacción (en folículos porcinos, tiroides de rata y tejido humano) utilizando como sustrato al ácido araquidónico, es la IL-o.
Específicamente podemos mencionar que la IL-o posee efectos inhibitorios sobre el crecimiento, como se ha demostrado en diferentes modelos estudiados.
En estudios in vitro la IL-o estimula la apoptosis en folículos tiroideos porcinos (Langer et al., 2003) e inhibe la proliferación celular en la línea FRTL-5 (Pisarev et al., 1992) e inhibe la acción proliferativa de la forscolina. Los autores sugirieron que la acción inhibitoria de la IL-o podría estar relacionada con una disminución en los niveles de AMPc o en un paso río abajo. Sin embargo en tirocitos de cerdo la IL-o inhibe la acción proliferativa del EGF y la formación de fosfatidil inositol estimulado por EGF, pero no tiene efecto sobre la acumulación de AMPc inducida por TSH (Dugrillon et al., 1990). En cortes de tiroides bovinas se ha demostrado que derivados iodados del ácido araquidónico, el 14-iodo-15-hydroxy-6-ácido eicosatrienoico (I-HO-A), su delta y omega lactona (lL-o y ILxo) reproducen los efectos inhibitorio del iodo tanto en lo que respecta a la captación (Chazenbalk et al., 1988), organificación (Chazenbalk et al., 1984) y generación de H20], (Krawiec el al., 1988). A nivel de la mernebrana celular Krawiec et al., (1991), han demostrado que el l-HO-A y su omega lactona (lLxo) reproducen los efectos inhibitorios del iodo sobre la captación de desoxiglucosa y la actividad de la Na+K+ ATPasa.
En ratas, la IL-o inhibe la generación del bocio inducida por MMI (Pisarev et al., 1988 y 1994), Y este efecto fue asociado con una disminución en los niveles de AMPc. Se ha demostrado que la TL-o no sólo previene el desarrollo del bocio cuando se administra simultáneamente con el boció-geno, sino que produce la involución del bocio preformado en ratas (Pisarev et al., 1988 y 1994).En los mismos estudios se demostró la ausencia de efectos tóxicos agudos, así como la eficacia terapéutica cuando el iodocompuesto es administrado tanto por vía oral como intraperitoneal. Hemos demostrado que la IL-o inhibe el crecimiento in vitro en células tiroideas normales, en una línea de cáncer folicular, en una línea de cáncer de colon y en una línea de rnelanoma, indicando que la tumorigénesis tiroidea podría deberse a la pérdida de la capacidad de organificar el iodo y de sintetizar lípidos iodados como la IL-o, compuestos capaces de inhibir la proliferación en distintas líneas celulares. Cabe aclarar que recientemente se ha demostrado también que la IL-o induce la apoptosis en células de cáncer de mama y en otros tipos de células cancerígenas (Arroyo-Helguera et al., 2006; Rosner et al., 2009)
Figura 1: Síntesis de IL-6

Sin embargo no todos los efectos del ioduro son reproducidos por la IL-8. Hemos demostrado, recientemente, que la expresión de genes específicos tiroideos es inhibida por el 1- pero no por la IL-8.
lodohexadecanal (IHD):
El lodohexadecanal es el principal lípido iodado sintetizado en la tiroides, su concentración es bastante mayor que la de IL-8. En 1990, Pereira et al. lo aislaron y purificaron. Es producido en tiroides de caballo, conejo y perro y sería el principal iodolípido sintetizado por la tiroides. Este compuesto reproduce los efectos del exceso de iodo sobre la producción de AMPc y de H202 (Pereira et al. 1990; Ohayon R 1994; Panneels I994a,b).
En membranas de tiroides porcinas el lHD inhibe a la NADPH oxidasa (hoy conocidas como DUOX1 y DUOX2, Ohayon et al, 1994). En cultivos de tiroides caninas el IHD disminuye los niveles de H202 (Panneels et al I994a) y disminuye la acumulación de AMPc inhibiendo directamente la actividad de la adenilato ciclasa (Panneels et al I994b). El IHD sería el mediador de dos importantes mecanismos regulatorios en la glándula tiroides: el efecto Wolff-Chaikoff y la inhibición de la adenilato ciclasa.
Recientemente hemos demostrado que este compuesto posee una actividad antibociógena, disminuyendo los niveles intra-celulares de cAMP, reduciendo el número de células y la altura del epitelio glandular. En estudios in vitro hemos observado que El IHD reguló negativamente el efecto de TSH sobre la proliferación de células FRTL-5 y la expresión de genes tiroideo específicos (NIS y Tg)
Rol del TGF-p en la autorregulación tíroídea
El TGF-p pertenece a una super familia de proteínas integradas por más de 35 citocinas que incluye a las activinas, inhi-binas, proteína morfogénica del hueso (BMP), hormona anti-müleriana y a las distintas isoformas de TGF-p, El TGF-p es sintetizado como precursor dimérico, secretado en forma lateote, esta forma inactiva es clivada siendo finalmente la región C-terminal la que conformará el péptido llamado TGF-p activo.
Existen cinco isoformas del TGF-p, en mamíferos se han des-cripto sólo las isoformas 1, 2 y 3.
Las señales de las tres isoformas del TGF-p son mediadas por dos receptores TpR-1I y TpR-1. De ambos receptores existen variantes, los cuales son productos generados por empalme splicing alternativo, Los receptores tipo 11 poseen actividad serina/treonina quinasas, son los primeros en unirse al ligando, para luego formar un complejo con el TpR-1 (Sosa-Garrocho & Silva, 2004).
Al formarse el complejo de receptores 1-11, el receptor tipo 11 fosforila al de tipo 1, esta fosforilación permite que el receptor tipo 1 se active y transmite las señales al interior celular por medio de la activación de las proteínas Smad, Las proteínas Smad comprenden tres subfamilias basándose en sus propiedades estructurales y funcionales: R-SMAD o Smad activadas por el receptor, que incluye las de tipo 2 y 3, Co SMAD o Smad4 son las que se asocian a las R-SMAD de modo de continuar. con la cascada de señales, y por último las Smad antagonistas o inhibitorias (1 SMAD), de las que forman parte Smad 6 y Smad 7 (Massagué, 1998; Moustakas et al., 2001; Bierie & Moses, 2006).
Se ha postulado la existencia de cascadas de señales inducidas por TGF-p, pero independientes de las proteínas Smad. (Seoane, 2006), como son por ejemplo la vía clásica de las Ras / MAPK / JNK la vía de 1P3K y Akt. Las funciones fisiológicas y fisiopatológicas del TGF-p son extensas y son múltiples los procesos fisiológicos y enfermedades en los que participa, El TGF-p afecta diversos procesos celulares, inhibe la proliferación celular pero también regula la diferenciación celular (Sosa-Garrocho & Silva, 2004). En cambio durante la progresión de la tumorogénesis el
no sólo pierde su acción antiproliferativa, sino que además se transforma en un factor oncogénico (Seoane, 2006). En las células tiroideas el del TGF-p es un potente inhibidor de la proliferación celular y de las funciones diferenciadas tiroideas.
En la tiroides, el ioduro estimula la síntesis (en cultivos de tiroides humanas) y secreción (en tiroides de oveja) de TGF-p, pero el efecto no se produce en presencia de MMI, indicando una vez más la existencia de algún iodocompuesto orgánico mediador. Además el TGF-pl reproduce algunos de los efectos inhibitorios del iodo sobre la función y proliferación tiroidea. Estos resultados llevaron a postular que este factor participaría en el mecanismo autorregulatorio. Gartner et al (1997) no encontraron efecto alguno de la lL-o sobre los niveles intracelulares de TGF-p.
Paradójicamente, el tratamiento con TSH o PTU, también estimula su síntesis (Morosini et al, 1996; Roger, 1996) y algunos autores han encontrado un aumento de TGF-p en el bocio. Este hallazgo se podría explicar como un mecanismo de balance que pretendería frenar la proliferación exagerada, o por una desregulación de la vía.
En folículos porcinos se observó inhibición del crecimiento e inducción de la apoptosis por parte del del TGF-p (Bechtner et al, 1999), también provoca una inhibición de la síntesis de Tg y TPO, inhibe la captación y de la organificación del iodo (Tsushirna et al., 1988; Delorme et al., 2002). En FRTL-5 el TGF- p inhibe a la Na+/K+ ATPasa (Pekary et al., 1997), la expresión de NIS estimulada por TSH (Kawaguchi et al., 1997), disminuye la captación de iodo y la síntesis de Tg (Coletta et al., 1989).
En resumen el TGF-p en células epiteliales normales posee acciones antiproliferaivas y proapoptóticas e inhibe las funciones diferenciadas en la glándula tiroides. En cambio en células tumorales esta respuesta se ha perdido y TGF-p estimula proliferación, invasión, angiogénesis y metástasis (Yingling et al., 2004). Conclusiones:
Los efectos autorregulatorios del iodo modulan la función y el crecimiento de la glándula tiroides a las diferentes señales de regulación. Sin embargo resta aclarar y profundizar en los estudios sobre los mecanismos e intermediarios a través de los cuales el iodo produce tantas y tan diversas acciones, donde parecería que el iodo actuaría a través de múltiples mecanismos, más que a través de una única vía de acción generalizada.
La importancia del mecanismo autoregulatorio está dada por diversas evidencias clínicas. Las acciones del iodo sobre el crecimiento tiroideo han sido uno de los efectos más estudiados.
Se ha demostrado una menor concentración de iodo en algunos nódulos y adenomas. La disminución en el contenido intratiroideo de iodo se explicaría por que en estas patologías es frecuente encontrar una disminución en la actividad de la TPO. En estas condiciones el iodo no se organifica y es liberado nuevamente a la circulación. Dado que la TPO también participaría en la biosíntesis de lípidos iodados, deberían estar disminuidos en estas patologías.
En regiones con una ingesta pobre en iodo se ha encontrado un aumento en la incidencia de cáncer folicular e indiferencia-dos mas agresivos (Pearce & Kendall-Taylor, 2005) y en las zonas de endemia en las que se implernentó la profilaxis con sal iodada se observó un cambio con predominio de la forma papilar (Harach et al., 2002). Más aún, en los estudios de Nishikawa et al. (2005), se ha reportado un aumento en la incidencia de cáncer de tiroides estimulado por DHPN en ratas que han recibido una dieta deficiente en iodo y rica en proteínas de saja (que inhiben a la TPO). Todos los resultados presentados y nuevos estudios son esenciales para poder avanzar y orientar una terapéutica en el caso de tumores tiroideos y de otras patologías asociadas y, es necesario por lo tanto, estudiar y aclarar el efecto de los iodo-lópidos sobre el crecimiento de distintas líneas de cáncer de tiroides humano. Nota: las figuras fueron modificadas de Panneels et al., 2009
REFERENCIAS BIBLIOGRÁFICAS
1. Arroyo-Helguera 0, Anguiano B, Delgado G, Aceves C. Uptake and antiproliferative effect of molecular iodine in the MCF-7 breast cancer cell line. Endocr Relat Cancer.2006, 13:1147-1158. [ Links ]
2. Bechtner G, Froschl H, Sachse A. Induction of apoptosis in porcine 1'01-lieJesbyTGF-~1 and EGF. Biochimie 1999,21:315-320.
3. Bierie B y Moses H. TGF-~. The molecular .Iekyll and Hyde of cancer. Nature reviews.2006,6:506-520.
4. Boeynaems Jlvl y Hubbard Wc. Transformation of arachidonic acid into an iodolactone by the rat thyroid. .I. B. C. 1980,255:9001-9004.
5. Boeynaems .1M, Pelster, D, Oates .lA and Hubbard Wc. Novel transformation of arachidonic acid by the rat thyroid in vitro. Biochirnica et Biophisica Acta 1981(c),665:623-627.
6. Boeynaems, .I.M., Reagan, D., Hubbard, W.c. Lactoperoxidase-cat-alyzed iodination of arachidonic acid: forrnation of macrolides. Lipids, 1981 (a) 16:246-249.
7. Boeynaems. J.M., Watson, .I.T., Oates, .I.A., Hubbard, w.c. lodination of docosahexaenoic acid by lactoperoxidase and thyroid gland in vitro: formation of an lodolactone. Lipids, 1981 (b), 16:323-327.
8. Bray G. Increased Sensitivity of the Thyroid in lodine-Depleted Rats to the Goitrogenic Effects of Thyrotropin. .I. Clin. Invest. 47: 1640-1647, 1968
9. Chazenbalk GD, Pisarev MA, Krawiec L, .Iuvenal G.I, Burton G, Valsecchi RM.ln vitro inhibitory effects of an iodinated derivative of arachidonic acid on calf thyroid.Acta Physiol Pharmacol Latinoam. 1984;34:367-373.
10. Chazenbalk, G.D., Valsecchi, R.M., Krawiec, L., Burton, G.. Juvenal, G.J., Monteagudo, E., Chester, H.A., Pisarev, M.A. Thyroid autoregula-tion inhibitory effects 01'iodinated derivatives of arachidonic acid in iodine metabolism. Prostaglandins. 1988,36:163-172.
11. Cochaux P, Van Sande .l, Swillens S, Dumont .lE. lodide-induced inhibi-tion of adenylate cyclase activity in horse and dog thyroid. European .Iournal of Bíochemistry. 1987,170:435-442.
12. Coletta G, Cirafaci AM, Di Carlo A. Dual effect of transforrning growth factor ~ on rat thyroid cells: inhibition of thyrotropin-induced prolifera-tion and reduction in thyroid-specific differentiatiom markers, Cáncer Res. 1989, 49:3457-3462.
13. Delange y Dunn. lodine Deficiency in The Thyroid, 9th Edition,LE
14. Braverman & RD Uitger,eds., Lippincott USA, 2005.. Delorrne , Re1l10nd C, Sartelet, H. TGF~I effects on functional activity of porcine thyroid cells cultured in suspension. J. Endoc. 2002,173:345-355.
15. Degroot, L..I. and Greer, M.A. Metabolisrn 1956, 5:682-696.
16. Dugrillon A, Bechtner G, Uedelhoven WM. Evidence that an iodolactone rnediates the inhibitory effect of iodine on thyroid cell proliferation but not on adenosine 3',5'-rnonophosphate formation. Endocrinology. 1990,127:337-343.
17. Dugrillon A, Uedelhoven WM, Pisarev MA. Identification of a 0-iodolactone in iodide treated hurnan goiter and its inhibitory effect on proliferation on hurnan thyroid follicles. Horrn. Met, Res. 1994,26:465-469.
18. Durnont JE, Lamy F, Roger PP. Physiological and pathological regulation of thyroid cell proliferation and differentiation by thyrotropin and other factors. Physiological Rev. 1992,72:667-697.
19. Eng PHK., Cardona GR, Fang SL. Escape frorn the acute Wolf-Chaikoff effect is associated with a decreased in thyroid sodium/iodide symporter (IS) expression. Endocrinology 1999; 140:3404.
20. Gartner R, Greil W, Dernharter R. Involvernent of cAMP, iodide and metabolites of arachidonic acid in the regulation of cell proliferation of isolated porcine follicles. Mol. Cell. Endocrinol. 1985,42:145.
21. Gartner R, Schopohl D, Schaefer S. Regulation of TGF-~Ir nRA expression in porcine thyroid follicles in vitro by growth factors, iodine or o-iodolactone. Thyroid. 1997, 4:633-640.
22. Harach H, Escalante D, Day E.Thyroid Cancer and Thyroiditis in Salta, Argentina: a 40 yr study in relation to iodine prophylaxis. Endocr. Pathol 2002,13:175-181.
23. Juvenal GJ, Pisarev MA, Kleiman de Pisarev DL, AltschulerN. Uptake, metabolisrn and action of triiodothyronine in calf-thyroid slices.Mol Cell Endocrinol. 1981,22:31-40
24. Kawaguchi A, Ikeda M, Endo T, Kogal T, Miyazaki A, Onaya T. Transforrning growth factor ~1 suppresses thyrotropin induced Na+/I-syrnporter rnessenger RNA and protein levels in FRTL-5 rat thyroid cells. Thyroid 1997, 7:789-794.
25. Krawiec L, Chazenbalk GD, Puntarulo, SA. The inhibition of PB1251 forrnation on calf thyroid caused by 14-iodo-1 5 hydroxy-eicosatrienoic acid is due to effect lo decreased H202 availability. Horm. Metab. Res. 1988,20:86.
26. Krawiec L, Chester HA, Bocanera LV, Pregliasco LB, Juvenal GJ, Piarev MA. Thyroid autoregulation: evidence for an action of iodoarachidonates and iodide at the cell rnernbrane level. Honnone and Metabolic Research 1991,23:321-325.
27. Langer R, Burzler C, Bechtner G, Gartner R.lnfluence of iodide and iodolactones on thyroid apoptosis. Evidence that apoptosis induced by iodide is mediated by iodolactones in intact porcine thyroid follicles. fu Clin Endocrinol Diabetes. 2003,111:325-329.
28. Laurent E, Mockel J, Takazawa K, Erneux C, Durnont JE. Stimulation of generation of inositol phosphates by carbamyJcholine and its inhibition by phorbol esters and iodide in dog thyroid cells. Biochernical Journal 1989; 263:795-801
29. Massagué J: TGF-~ signal transduction. Annu. Rev. Biochem. 1998,67:753-791.
30. Morosini Pp, Taccaliti A, Arnaldi G, Simonella G, Petrelli Md, Mancini V, Montironi R, Scarpelli M, Diamanti L, Mantero F. Enhanced Expression Of Transforming Growth Factor Beta 1 In Rat Thyroid Iyperplasia Is Thyrotropin Induced And Time Dependent. Eur. .l. Endocrinol. 1996,134:373-378.
31. Moustakas A, Souchelnytski S, I-Ieldin CH: Smad regulation in TGF-~ signal transduction. J. Cell Sci.2001, 114: 4359-4369.
32. Nishikawa A, Ikeda T, Son HY, Okazaki K, lmazawa T, Umemura T, Kimura S, Hirose M. Toxicol Sci. Pronounced synergistic promotion of N-bis(2-hydroxypropyl)nitrosamine-initiated thyroid tumorigenesis in rats treated with excess soybean and iodine-deficient diets. Toxicol Sci. 2005,86:258-263.
33. Ohayon R, Boeynaerns JM, Braekman JC, Van den Bergen H, Gorin Y, Virion A. Inhibition of thyroid ADPH oxidase by 2-iodohexadecanal in a cell-free systern. Mol. Cell Endocrinol. 1994, 99:133-141.
34. Panneels V, Juvenal G, Boeynaems J.M, Dumont JE, J. Van Sande, lodide Effects on The Thyroid: Biochemical, Physiological, Phannacological And Clinical Effects Of lodide In The Thyroid. Cornprehensive Handbook on lodine: utritional , Endocrine and Pathological Aspects. V. R. Preedy, G. . Burrow, R. Watson, eds, Oxford:Academic Press,2009, pp. 303-314
35. Panneels V, Macours P, Van der Bengen H. Biosynthesis and metabolisrn of 2-iodohexadecanal in cultured dog thyroid cells. J. Biol Chern. 1996,271:23006-23014.
36. Panneels, v., Van den Bergen 1-1., Jacoby, C.; Braekrnan, J.c., Van Sande, J., Dumont, J.E., Boeynaems, J.M. Inhibition of H2O2 production by iodoaldehydes in cultured dog thyroid cells. Mol. Cell Endocrinol. 1994 (a),102:167-176.
37. Panneels, v., Van Sande, J., Van den Bergen H.. Jacoby, c., Braekrnan, .l.C.; Dumont, J.E., Boeynaems, J.M. Inhibition of human thyroid adeny-Iyl cyclase by 2-iodoaldehydes. Mol. Cell Endocrinol. 1994 (b),106,:41-50.
38. Pearce & Kendall-Taylor, Genetic Factor in Thyroid Diseases in The Thyroid, 9th Edition,LE Braverrnan & RD Uitger,eds., Lippincott USA, 2005.
39. Pekary AE, Levin SR, Johnson DG, Berg L, Hershman JM. Tumor necrosis factor-alpha and rransforming growth factor-beta 1 inhibit the expression and activity of a+/K+-ATPase in FRTL-5 rat thyroid cells. J.lnterf.Cytokine Res. 1997,17:185-195.
40. Pereira A, Braekman JC, Durnont JE. Identification of a major iodolipid frorn the horse thyroid gland as 2-iodohexadecanal. J. Biol. Chem. 1990,265:17018-17025.
41. Pisarev MA, Utiger RD, Salvaneschi JP, Altschuler N, DeGroot LJ. Serum TSI-I and Thyroxine in Goitrous Subjects in Argentina. J Clin Endocr 1970,30:680-681.
42. Pisarev MA, Bocanera LV, Chester I-IA, Krawiec L, JuvenalGJ. Effect of iodoarachidonates on thyroid FRTL-5 cells growth. Horm. Metab. Res. 1992,24: 558.
43. Pisarev MA, Chazenbalk GD, Valsecci RM. Thyroid autoregulation. Inhibition of goiter growth and ofcAMP fonnation in rat thyroid by iod-inated derivatives of arachidonic acid, J. Endocrinol. Invest 1988,1:669-674.
44. Pisarev MA, Gartner R Autoregulatory action of iodine, in The Thyroid, 8th Edilion,LE Braverman & RD Uitger,eds., Lippincott USA, 2000. pages. 85-90.
45. Pisarev MA, Krawiec L, Juvenal GJ. Studies on the goiter inhibiting effects of iodolactones. Eur. J. Pharmacol, 1994,258:33-37.
46. Pisarev MA. Thyroid auioregulation. J. Endocrinol. Invest. 1985,8: 475-484.
47. Pregliasco L, Bocanera, Krawiec, Pisarev M, Juvenal G. Effects of iodide on thyroglobulin biosynthesis in FRTL5 cells. Thyroid 1996,6:319-323.
48. Roger PP: Thyrotropin-dependenl transforming growth factor beta expression in thyroid gland. Eur. J. Endocrinol. 1996,134: 269-271.
49. Rognoni, J.B., Penel, c., Ducret, F. Respectives roles of circulatin T4 and T3 in control of TSH secretion in severely iodide-deficient rats. Acta Endocrinol (Copenh), 1984,105:40-48.
50. Rousset, Dunn. In De Groot, LJ: "Thyroid disease manager", Chapter 2: Thyroid Hormone Synthesis and Secretion, 2004
51. Seoane J. Escaping from the TGFbela anti-proliferative control. Carcinogenesis. 2006,27:2148-2156.
52. Smerdely P, Pitsiavas V, Boyages S. The G2M arrest caused by iodide is unrelated to the effects of iodide at Adenylate Cyclase.Thyroid, 1995,5:325-330.
53. Sosa-Garrocho M, Silva M. El factor de crecimiento transfonnante ~: Funciones y Vías de Transducción. REB. 2004,23:3-11.
54. Tsushirna T, Arai M, Motoyasu S. Effects of TGF-~ on DNA sinthesis an iodine metabolism in porcine thyroid cells in culture. Endocrinology 1998,123: 1187-1193.
55. Van Sande J, Grenier G, Willems C, Dumont JE. Inhibition by iodide of the activation of the thyroid cyclic 3',5'-AMP system. Endocrinology 1975,96:781-786.
56. Wolff J, Chaikoff 1L, Goldberg RC. The temporary nature of the inhibitory action of excess iodide on organic iodide synthesis in normal thyroid. Endocrinology. 1949; 45:504.
57. Wynn JO.. Components of the serum protein-bound iodine following adrninistration of I -labeled hog thyroglobulin. J. Clin. Endocrinol. Metab. 1961,21: 1572.
58. Yingling JM, Blanchard KL, Sawyer JS. Development of TGF-beta sig-nalling inhibitors for cancer therapy. al. Rev. Drug Discov. 2004,3(12):1O11-1022. Correspondencia: Guillermo Juvenal División Bioquírnica Nuclear
Comisión acional de Energía Atómica
Av. Libertador 8250
1429 Buenos Aires, Argentina
juvenal@ cnea.gov.ar