








Servicios Personalizados
Articulo
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
Links relacionados
 Citado por SciELO
Citado por SciELO
 Similares en SciELO
Similares en SciELO
Bookmark
Archivos Bolivianos de Medicina
versión impresa ISSN 0004-0525
Arch.Boliv.Med. v.13 n.81 Sucre 2009
CASOS CLÍNICOS
Holoprosencefalia
* Dra. Doris Alina Rodríguez C. Médico - Pediatra. Docente titular Cátedra de Pediatría U.M.R.P.S.X.Ch (Sucre – Bolivia)
** Dr. José Luís Chavarria R. Médico Pediatra – Neonatólogo. Docente titular Cátedra de Pediatría U.M.R.P.S.X.Ch (Sucre – Bolivia)
*** Dr. Gary Coila F. Jefe de Servicio de Pediatría - Neonatología “Hospital Universitario” A.V.B. (Sucre - Bolivia)
**** Dr. Javier Caihuara LL. Médico – Residente III de Gineco-obstetricia: Hospital de la Mujer “Jaime Sánchez Pórcel”
PALABRAS CLAVE.-
• Recién nacido Cíclopes
• Holoprosencefalia
INTRODUCCIÓN
LA HOLOPROSENCEFALÍA
Es un trastorno caracterizado por la ausencia del desarrollo del prosencéfalo (el lóbulo frontal del cerebro del embrión). Durante el desarrollo normal se forma el lóbulo frontal y la cara comienza a desarrollarse en la quinta y sexta semana del embarazo. La holoprosencefalia es causada por la falta de división del lóbulo frontal del cerebro del embrión para formar los hemisferios cerebrales bilaterales (las mitades izquierda y derecha del cerebro), causando defectos en el desarrollo de la cara y en la estructura y el funcionamiento del cerebro.
Existen tres clases de holoprosencefalia. La holoprosencefalia alobar es el tipo más grave, en la cual el cerebro no logra separarse y se asocia generalmente a anomalías faciales severas. La holoprosencefalia semilobar, en la cual los hemisferios del cerebro tienen una leve tendencia a separarse, constituye una forma intermedia de la enfermedad. La holoprosencefalia lobar, en la cual existe una evidencia considerable de separación de los hemisferios del cerebro, es la forma menos grave. En algunos casos de holoprosencefalia lobar, el cerebro del paciente puede ser casi normal.
La holoprosencefalia, denominada anteriormente como arinencefalia, consiste en una gama de defectos o malformaciones del cerebro y de la cara. En el extremo más grave de este espectro se encuentran los casos que involucran malformaciones serias del cerebro, malformaciones tan graves que son incompatibles con la vida y a menudo causan la muerte intrauterina espontánea. En el otro extremo del espectro están los individuos con los defectos faciales -que pueden afectar los ojos, la nariz y el labio superior y el desarrollo normal o casi normal del cerebro. Pueden ocurrir convulsiones o retraso mental.
El más grave de los defectos (o anomalías) faciales es la ciclopia, caracterizado por el desarrollo de un solo ojo, que se ubica generalmente en el área ocupada normalmente por la raíz de la nariz, y la ausencia de la nariz o una nariz en la forma de una probóscide (un apéndice tubular) situada por encima del ojo.
La etmocefalia es la anomalía facial menos común. Consiste en una probóscide que separa ojos muy juntos, ausencia de la nariz y microftalmia (tamaño anormalmente pequeño de uno o ambos ojos).
La cebocefalia es otra anomalía facial caracterizada por una nariz pequeña y aplastada con un solo orificio nasal situada debajo de unos ojos subdesarrollados y muy juntos. La anomalía facial menos grave es el labio leporino, también llamado agenesia premaxilar.
Aunque las causas de la mayoría de los casos de holoprosencefalia siguen siendo desconocidas, los investigadores saben que aproximadamente la mitad de todos los casos se deben a causas cromosómicas (de los cromosomas). Las anomalías cromosómicas, tales como el síndrome de Patau (triso mía 13) y el síndrome de Edwards (triso mía 18) se han podido asociar con la holoprosencefalia. Los hijos de madres diabéticas tienen un riesgo mayor de padecer el trastorno.
No existe tratamiento para la holoprosencefalia y el pronóstico para los individuos que la padecen es pobre. La mayoría de los que sobreviven no muestran signos de desarrollo significativos. Para los niños que sobreviven, el tratamiento es sintomático (es decir, alivia sólo los síntomas y no las causas del trastorno). Es posible que una mejora en el monitoreo de embarazos de madres diabéticas pueda ayudar a prevenir la holoprosencefalia. No obstante, no existen medios de prevención primaria.
DESCRIPCIÓN DEL CASO.-
Se trata de una paciente que acude al servicio de emergencias obstétricas del hospital de la mujer de 18 años de edad con el antecedente de dinámica uterina y dilatación de 9 cm, por lo que ingresa inmediatamente a sala de partos. Con los siguientes diagnósticos obstétricos:
- embarazo de 37 semanas por examen físico.
- trabajo de parto
Se obtiene un producto único vivo, por examen físico corresponde a 36 semanas, de gestación: sexo femenino, apgar 3 al minuto y 3 a los 5 minutos, peso = 2800 grs., talla = 49 cm., perímetro cefálico = 29.5 cm.
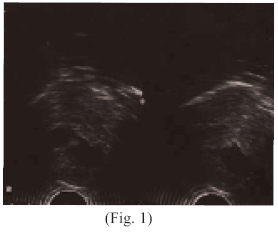
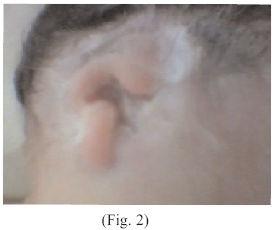
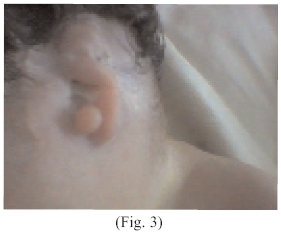
Al examen físico neonatal; cráneo microcéfalo, con un perímetro cefálico disminuido, ecograficamente se observan anencefalia (Fig. 1), cabellos delgados, finos mal implantados, con zonas alopecicas en región occipital de más o menos 1 cm, en número de uno. Orejas mal implantadas, pabellones auriculares aplásicos, o esbozos, (Fig. 2 y 3)
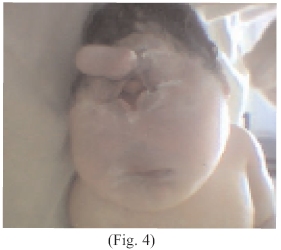
Fascies: pequeña con ciclopia con un ojo de 2 cm de diámetro emergiendo en el lugar de la raíz de la nariz a nivel de la frente. (Fig. 4)
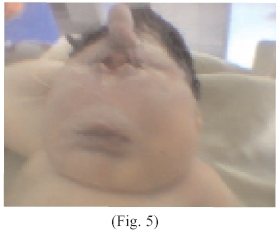
Se observa una formación cilíndrica de diámetro de 1 cm., y largo 2,5 cm. con cuya parte final presentaba una depresión simulando una fosa nasal, en forma de una probóscide (apéndice tubular), ausencia de párpados superiores e inferiores, ausencia fosas nasales y de la nariz, (arrinia) (Fig. 5) labios y cavidad bucal con características físicas aparentemente normales, cuello engrosado con aumento de la masa muscular, corazón: ruidos cardiacos rítmicos hipofonéticos rítmicos, con presencia de soplo sistólico.
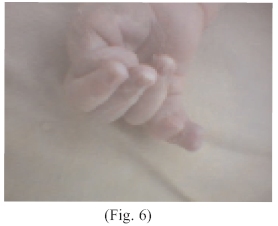
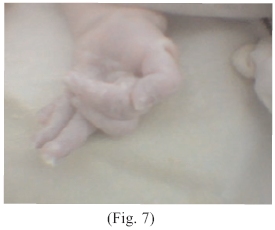
Extremidades superiores con hexadactilia, pulgar retroflexible, yuxtaposición de dedos anular y meñique (Fig.: 6 y 7) resto del cuerpo supuestamente normal.
Llegando a fallecer a los 30 minutos de vida.
En cuanto a los antecedentes patológicos de la madre: es un producto de segundo embarazo, el primer hijo es normal, no refiere enfermedades padecidas durante la gestación, ni haber ingerido medicamento alguno, el único dato importante su alimentación en cuanto a hortalizas y legumbres, proviene de la zona de Quirpinchaca, lugar donde drenan las aguas no potables de la zona urbana de la ciudad de Sucre.
DISCUSION
En los últimos años se observa con preocupación una mayor incidencia de malformaciones congénitas en especial del sistema nervioso central. Entre los factores desencadenantes se encuentran los genéticos y los factores exógenos a los que se ve expuesta la población en etapa de gestación. La falta de control prenatal no permite un diagnóstico de riesgo gestacional oportuno, lo que permitiría tomar las acciones tanto en aspectos preventivos como terapéuticos.
RECOMENDACIONES
Al ser la gestación una etapa de riesgo tanto para la madre como para el niño, se requieren tomar acciones urgentes con la finalidad de prevenir las complicaciones del embarazo, promover un buen estado de salud en el binomio madre-niño y detectar en forma precoz los problemas que se presentan durante la gestación. Es por ello que todas las instituciones de salud y todo su personal deberán priorizar la atención de este grupo poblacional y orientar todos los recursos económicos, técnicos y logísticos para lograr evitar las situaciones que presentamos anteriormente.
BIBLIOGRAFÍA
1. Zol B, Wolf J, Lensing-Hebben D, Pruggmayer M, Torpe B. Trisomy 13 with an 11-years survival. Clin Genet 1993; 43 (1): 46-50.
[ Links ]2. Hirschhorn K. Enfermedades prenatales. En: Berhrman RE. Nelson. Tratado de Pedriatía. 14ª ed. Madrid: WB Saunders Company, 1992; 319-66.
3. Patau K, Smith DW, Therman E, Inhorn SL, Wagner HP. Multiple congenital anomaly caused by an extra autosome. Lancet 1960; 1: 790-3.
[ Links ]4. Edwards JH, Harnden DG, Cameron AH, Crosse VN, Wolff OH. A newtrisomic syndrome. Lancet 1960; 1: 787-9.
[ Links ]5. Huether CA, Martin RL, Stoppelman SM, D´souza S, Bishop JK, Torfs CP, et al. Sex ratios in fetuses and liveborn infants with autosomal aneuploidy. Am J Med Genet 1996; 63 (3): 492-500.
[ Links ]6. German J. Aspectos citogenéticos de la enfermedad humana. En: Fauci AS, Braunwald E, Isselbacher KJ, Wilson JD, Martin JB, Kasper DL, et al. Harrison. Principios de Medicina Interna. 14ª ed. Madrid: McGraw
[ Links ]