








Servicios Personalizados
Articulo
 Articulo en PDF
Articulo en PDF Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
Links relacionados
 Citado por SciELO
Citado por SciELO
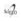 Similares en SciELO
Similares en SciELO
Bookmark
Revista de Investigación e Información en Salud
versión impresa ISSN 2075-6194
Rev. Inv. Inf. Salud v.10 n.24 Cochabamba 2015
ARTÍCULO CIENTÍFICO
Pesquisaje prenatal de hemoglobinopatías ss y cc: 25 años de experiencia en la Provincia de Villa Clara, Cuba
Prenatal screening of hemoglobinopathies ss and cc: 25 years of experience in the province of Villa Clara, Cuba
Dr. Noel Taboada Lugo 1 Ing. Melba Gómez Rojo 2 Dra. Ana E. Algora Hernández 3 Dra. Gretsy Arcas Ermeso 4 Dra. María D. Noa Machado 5 Dra.Manuela Herrera Martínez 6
1. Máster en Atención Integral al niño. Doctor en Medicina. Especialista de Primer Grado en Medicina General Integral. Especialista de Primer y Segundo Grado en Genética Clínica. Centro Provincial de Genética Médica. Villa Clara, Cuba. E-mail: taboada@capiro.cvl.sld.cu
2. Ingeniera Química. Jefa del Laboratorio de Hemoglobinopatías. Centro Provincial de Genética Médica. Villa Clara. E-Mail: melbar@capiro.vcl.sld.cu
3. Doctora en Medicina. Especialista de Primer y Segundo Grado en Genética Clínica. Centro Provincial de Genética Médica. Villa Clara. E-Mail: algora @capiro.vcl.sld.cu
4. Doctora en Medicina. Especialista de Primer y Segundo Grado en Genética Clínica. Centro Provincial de Genética Médica. Villa Clara. E-Mail: gar-cas@capiro.vcl.sld.cu
5. Máster en Asesoramiento Genético. Doctora en Medicina. Especialista de Primer Grado en Medicina General Integral. Centro Provincial de Genética Médica. Villa Clara. E-Mail: noamachado@infomed.vcl.sld.cu
6. Doctora en Ciencias Médicas. Especialista de Primer Grado y Segundo Grado en Genética Clínica. Universidad de Ciencias Médicas "Serafín Ruíz de Zárate" Villa Clara. E-Mail: manuelah@iscm.vcl.sld.cu
Institución que auspicia el trabajo: Centro Provincial de Genética Médica Villa Clara, Cuba.
Fecha de recepción: 22. 04.2015 | Fecha de aprobación: 14.05.2015
RESUMEN
Hemoglobinopatías es el término médico que designa a un grupo de trastornos que afectan a las células rojas de las sangre. El estado homoci-goto para la hemoglobina S es la forma más común y severa, aunque también se incluyen heterocigotos compuestos para la hemoglobina S yC.
Se realizó un estudio observacional, descriptivo y transversal donde se exponen los resultados de 25 años del programa de pesquisaje prenatal de hemoglobinopatías SS y SC en la provincia Villa Clara, Cuba, en el período comprendido de 1988 a 2012.
Se diagnosticaron 4 660 embarazadas portadoras de estas hemoglobinopatías y se estudiaron 4 193 cónyuges. Se identificaron 144 parejas de alto riesgo y 126 de ellas aceptaron procedimientos de diagnóstico prenatal. Se diagnosticaron 24 fetos afectados, de los cuales en 17 casos la pareja decidió la interrupción electiva de la gestación. La tasa de prevalencia ajustada de estas hemoglobinopatías en Villa Clara durante el periodo estudiado fue de 0,95 por 10 000 nacidos vivos.
Palabras Clave: Hemoglobinopatías, anemia fal-ciforme, Hemoglobina S, Hemoglobina C.
ABSTRACT
Hemoglobinopathies is the medical term that describes a group of disorders affecting the red blood cells. The homozygous state for hemoglobin S is the most common and severe form, although compound heterozygotes for hemoglobin S and C are included also. An observational, descriptive
and transversal study was performed, which displays the results of 25 years of the prenatal screening program of hemoglobinopathies SS and SC in the province of Villa Clara, Cuba, during the period 1988-2012.
4 660 pregnant women were diagnosed with these hemoglobinopathies and 4 193 husbands were studied. We identified 144 high-risk partners and 126 of them agreed to prenatal diagnostic procedures. 24 affected fetuses were diagnosed, of which in 17 cases the couple decided to elective termination of pregnancy. The adjusted prevalence rate of these hemoglobinopathies in Villa Clara during the studied period was 0,95 per 10 000 live births.
Keywords. Hemoglobinopathies, Sickle cell disease, Hemoglobin S, Hemoglobin C.
INTRODUCCIÓN
La hemoglobina es una proteína que es transportada dentro de los glóbulos rojos en la sangre y es la responsable del transporte de oxígeno, tanto en humanos como en otros vertebrados. La molécula de hemoglobina (Hb) tiene una estructura básica de cuatro cadenas polipeptídicas llamadas globi-nas, cada una de las cuales presenta un grupo prostético hemo que contiene hierro (1).
En humanos, el tetrámero está formado por dos subunidades tipo alfa (j) y dos tipo beta f ) codificadas por bloques de genes localizados en cromosomas diferentes. Los genes tipo alfa tienen su loci en el brazo corto del cromosoma 16 (16p13.3) (#OMIM 141800), mientras que los genes tipo beta se ubican en el brazo corto del cromosoma 11 (11p15.4) (#OMIM 141900) (1) (2).
Ambos grupos de genes se organizan en el Ácido desoxirribonucleico (ADN) desde el extremo 5la 3ke la misma manera como se expresan durante el desarrollo embrión-adulto. Es así como un individuo durante la ontogenia presenta diferentes tipos de Hb que responden a la disponibilidad del oxígeno en los ambientes en los cuales le corresponde desarrollarse. Primero, se activan las hemoglobinas embrionarias; la Hb fetal comienza a ser el tetrámero dominante durante la vida fetal y justo antes del nacimiento la Hb fetal es reemplazada por las hemoglobinas adultas A1 yA2 (1).
Las moléculas de Hb pueden sufrir alteraciones por mutaciones ocurridas en los genes que codifican para las globinas, dando origen a las hemo-globinopatías, los desórdenes hereditarios más comunes que afectan al hombre (1) (3). Cuando la mutación afecta la secuencia de aminoácidos de la globina se forman las variantes estructurales de la Hb. Las más distribuidas en las poblaciones humanas son las hemoglobinas S, C, D y E, siendo la Hb S la que presenta las consecuencias clínicas más graves (1) (4).
Se denomina hemoglobinopatía al defecto de carácter hereditario, que tiene como consecuencia una estructura anormal en una de las cadenas de las globina de la molécula de Hb. Sin embargo, suele reservarse el término Hemoglobinopatías para las anomalías de la Hb producidas por el simple cambio de un aminoácido en una de las cadenas de globina; el término talasemias se reserva para las hemoglobinopatías debidas a la falta de síntesis, total o parcial, de una cadena completa de globina (4).
No todas las hemoglobinopatías producen manifestaciones clínicas, sin embargo, cuando éstas ocurren son variadas, presentándose en forma asintomática, leve o severa, como es el caso de la Hemoglobinopatía S homocigota, que presenta cuadros de anemia hemolítica crónica, originada por una mutación puntual en el codón 6 de la cadena tipok (Glut > Val). Esta modificación de la carga superficial de la hemoglobina disminuye la solubilidad de la Hb, especialmente en su estado reducido (deoxi-Hb), y facilita la formación de agregados fibrilares o polímero de molécula de Hb que alteran profundamente la morfología eritroci-taria y aumentan su rigidez (1) (5).
En su estado homocigoto produce la anemia de células falciformes, anemia drepanocítica, hemoglobinopatía S o más comúnmente Sicklemia (anglicismo por Sickle cell anemia), llamada así porque las células rojas afectadas toman la forma de hoz. Es una enfermedad hereditaria, multisistémica, asociada con episodios agudos y daño progresivo de diversos órganos. (# OMIM 603903) (1) (4) (6).
Le sigue en frecuencia la hemoglobina C, la cual es el resultado de una mutación en el mismo condón 6 de la cadena k (Glut > Lys). Las personas que tienen un alelo A y otro S, son denominados portadores sanos de Hemoglobina S (HbAS) y las que tienen un alelo A y otro C, son denominados portadores sanos de hemoglobina C (HbAC). Además, se utiliza el término rasgo falciforme, en el primer caso; y el de rasgo de la hemoglobina C, en el segundo. Los portadores sanos son asinto-máticos en condiciones normales, pero tienen una probabilidad de 50% de trasmitir a sus hijos cada uno de sus alelos para este gen (1) (7).
Según la Organización Mundial de la Salud (OMS), los desórdenes de la hemoglobina representan un problema de salud pública en 71% de 229 países. Cada año nacen más de 300 000 niños afectados, 83% con anemia de células falci-formes y 17% con talasemia. Las hemoglobinopa-tías, explican aproximadamente 3,4% de las muertes en niños menores de cinco años de edad (8).
Con el propósito de lograr la detección temprana de parejas de alto riesgo de tener hijos con hemo-globinopatías SS y CC en una edad gestacional lo más temprana posible, se estableció en los años 1991-1992 en el sistema nacional de salud de Cuba que el pesquisaje de hemoglobinas anormales en gestantes se hiciera en la captación del embarazo en el nivel primario de salud. Al identificarse una gestante portadora de hemoglobinas AS, AC o SC se cita a la pareja a los centros municipales de Genética Comunitaria, donde es valorada y asesorada por los másteres en Asesoramiento Genético y se indica el estudio al cónyuge, de ser también este portador de una de estas variantes de hemoglobina son remitidos a los Centros Provinciales de Genética Médica.
El Programa de Prevención de Anemia Falciforme, existente en la República de Cuba desde el año 1983 y que desde el año 1988 se extendió a todo el país, brinda a estas parejas con alto riesgo de tener hijos afectados con hemoglobinopatías SS y SC, la opción de realizar un diagnóstico prenatal (DPN), de detectarse un feto afectado por estas hemoglobinopatías se le brinda asesoramiento genético por parte de los especialistas en Genética Clínica, en los centros provinciales de Genética Médica, y la pareja puede decidir la interrupción electiva de la gestación. En todos los casos en que la pareja decida continuar la gestación, los neonatos con diagnóstico confirmado al nacer son enviados a las consultas de Hematología donde los padres reciben información adicional sobre la enfermedad y se inicia precozmente el tratamiento y la prevención de las complicaciones. Ello forma parte de otro programa nacional, el de Atención Integral al Paciente con Sicklemia, creado en 1990, que tiene como objetivo unificar criterios diagnósticos y terapéuticos de la enfermedad en todo el país y con el que se ha logrado disminuir la mortalidad en niños y adultos y mejorar su calidad de vida (7) (9).
La provincia de Villa Clara se ubica en la región central de la República de Cuba, ocupa el quinto lugar en extensión y población entre las provincias con 8 413,13 kms2, representando el 7,7 % de la superficie total del país. Según datos publicados por la Oficina Nacional de Estadísticas, representa el 7,1 % de la población del país con 803 562 habitantes, para una densidad de población de 95,5 habitantes por km2, (10) cuenta con 13 centros municipales de Genética Comunitaria y un Centro provincial de Genética Médica, donde laboran cinco especialistas en Genética Clínica.
Los objetivos de la presente investigación fueron: estimar la prevalencia ajustada de las hemoglobinopatías SS y SC diagnosticadas prenatalmente en la provincia Villa Clara, en el período comprendido de 1988 a 2012; y determinar la proporción de gestantes portadoras de Hb SS y SC del total de gestantes captadas por años, la proporción de cónyuges estudiados, el número de diagnósticos prenatales realizados, y el número de fetos afectados.
MATERIALES Y MÉTODOS
Se realizó un estudio observacional, descriptivo y transversal en el período comprendido de 1988 a 2012, los datos fueron obtenidos de los registros y archivos de la consulta de Hemoglobinopatías del Centro Provincial de Genética Médica de Villa Clara; Cuba.
La tasa de prevalencia ajustada al nacimiento de hemoglobinopatías SS y SC se calculó mediante la siguiente fórmula:

En el total de afectados se incluyó a todos los recién nacidos vivos afectados por hemoglobino-patías SS y SC, el total de mortinatos afectados y el total de interrupciones electivas de la gestación de fetos afectados.
Ya que el número de afectados se incrementa cuando se tienen en cuenta las interrupciones electivas de la gestación, es necesario considerar un denominador apropiado para realizar los cálculos de frecuencia. El denominador tradicional es el total de nacidos vivos y nacidos muertos. Lógicamente sería excluido gran número de abortos de menor peso, por lo que muchos registros de malformaciones y enfermedades genéticas en el mundo han decidido añadir las interrupciones electivas de la gestación por causas genéticas (11).
Así, el total de nacimientos ajustado se calculó sumando el total de recién nacidos vivos, el total de mortinatos afectados y el total de interrupciones electivas de la gestación de fetos afectados.
El número total de nacidos vivos en la provincia de Villa Clara en el periodo comprendió de 1988 al 2012 fue de 253 794.
RESULTADOS
En el periodo de 25 años comprendido de 1988 a 2012 se estudiaron un total de 247 984 gestantes en la provincia de Villa Clara, mediante estudio de electroforesis de hemoglobina indicada por su médico de familia en la consulta de captación del embarazo.
El total de gestantes portadoras de hemoglobino-patías (Hb S y/o C) fue de 4 660, que representan el 1,88% del total de estudiadas, solo en el año 1991 se diagnosticó menos de un caso por cada 100 embarazadas (Figura N°1) y se estudiaron 4 193 cónyuges (89,99%).
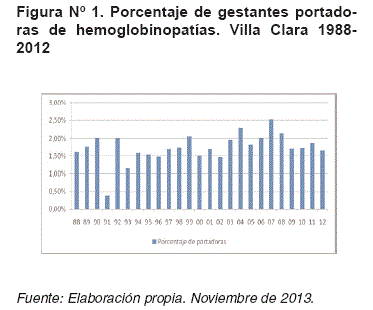
La Figura N°2 muestra la proporción de gestantes portadoras de hemoglobinopatías y cónyuges estudiados por años.
Se diagnosticaron 144 parejas de alto riesgo y de ellas a 126 parejas se les realizó DPN, lo que representa el 87,50%. A 18 parejas no se les realizó el proceder por las causas que se exponen: en 8 casos por avanzada edad gestacional, en 6 por abortos espontáneos donde no se precisó el tipo de Hb, en 3 casos por traslados de la embarazada de domicilio a otra provincia o país, y solamente en 1 caso porque la pareja no brindó el consentimiento informado para su realización.
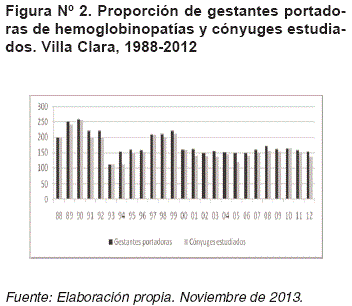
A través del DPN realizado por técnicas de biología molecular, se diagnosticaron 24 fetos afectados (19,01%), de los cuales en 17 casos (70,83%) la pareja decidió la interrupción de la gestación una vez que se les brindó asesoramiento genético (Figura N°3).
En 7 recién nacidos vivos se diagnosticó hemo-globinopatías SS o SC por lo que la tasa de pre-valencia ajustada de estas hemoglobinopatías en la provincia de Villa Clara durante el periodo estudiado fue de 0,95 por 10 000 nacidos vivos.
DISCUSIÓN
La frecuencia del estado de portador de hemoglobinopatías Hb AS y AC en embarazadas de la provincia de Villa Clara es aproximadamente 2%. De acuerdo a informes de la OMS, aproximadamente un 5% de la población mundial es portadora sana de un gen de la drepanocitosis, e incluso esta cifra puede alcanzar el 25% en algunas regiones (12).
La incidencia de portadores en la población cubana es de 3,085 por cada 100 habitantes, con grandes variaciones en dependencia del fenotipo color de la piel, así en las personas con color de la piel negra la incidencia es 10 veces superior (13,25 por cada 100 habitantes) y 0,65 en personas con piel blanca. La prevalencia de portadoras de Hb C es de 0,7%. Se calcula que el número de portadores de hemoglobina (Hb) S en Cuba es de alrededor de 300 000, y el número de enfermos aproximadamente de 4 000, distribuidos en todo el país, con un mayor número de enfermos en la ciudad de La Habana y en las provincias del sur de la región oriental (12) (13) (14).
Se estima que más de 2 millones de norteamericanos son portadores de sicklemia y que aproximadamente 70 000 presentan la enfermedad. Existe una percepción errada de que la enfermedad solo afecta a personas con ancestros africanos, sin embargo, esta enfermedad puede afectar a personas de cualquier grupo étnico y color de la piel (15).
La enfermedad está ampliamente difundida por Latinoamérica, en especial, el Caribe insular y países como Colombia, Venezuela y Brasil; se extiende por el Mediterráneo y es tan endémica en África como lo es la malaria, con la que comparte las zonas de distribución geográfica. De hecho, la teoría prevaleciente acepta que la mutación responsable de la dolencia puede ser la respuesta evolutiva a la presencia de la malaria, al conferir a los eritrocitos protección natural a la infección por el Plasmodium falciparum, uno de los parásitos más patógenos del phyllum apicomplexa y la especie responsable de la forma más severa de malaria humana, aunque los mecanismos para esta protección no están aún bien dilucidados, es probable que estén involucrados mecanismos genéticos y mediados por la inmunidad (16) (20).
La imposibilidad de realizar el estudio de los cónyuges en todos los casos, es un aspecto que contribuye a la no detección de las parejas de alto riesgo. En el territorio villaclareño, de las 4 660 gestantes portadoras, se desconoció el genotipo de sus parejas en 467 casos, con mayor incidencia en los años 1995 y 2005, de modo que 10,02% de estas parejas hubieran podido tener un hijo afectado sin que hubieran podido acceder al DPN. A nuestro juicio el comportamiento de este indicador se debe no solo a la inestabilidad de las parejas, el traslado hacia otros lugares de residencia por motivos laborales o sociales y el no reconocimiento de la paternidad; sino también, a la falta de información que tiene la población sobre la necesidad del estudio de ambos miembros de la pareja para la estimación del riesgo y poder ofrecer el diagnóstico prenatal de la enfermedad.
Estimamos conveniente para disminuir el número de parejas en que no se estudia el cónyuge, continuar estimulando el estudio preconcepcional y de ambos miembros de la pareja al momento de la captación del embarazo en el nivel de atención primario, así como trabajar por elevar los conocimientos de la población en este sentido.
El porcentaje de esposos estudiados (89,98%) durante el periodo de estudio en la provincia de Villa Clara resulta superior a lo observado a nivel nacional en Cuba hasta el año 2009 (82,5%), así como en otras provincias del país (Pinar del Río 2007 (78,9 %) y Holguín 2008 (83,3 %), sin embargo, en el municipio pinareño de Minas de Matahambre se logró estudiar al 95% de los cónyuges (21).
Al mejorar los indicadores de detección temprana de las parejas de alto riesgo, se debe elevar, en consecuencia, el número y proporción de las parejas que se realizan DPN. La detección temprana de la condición de la pareja de alto riesgo, ofrece la ventaja de encontrar el momento más apropiado para hacer la toma de la muestra, teniendo en cuenta el estado de salud de la gestante y permite repetir este proceder obstétrico en caso de ser necesario.
El comportamiento de las parejas de alto riesgo frente a la opción de DPN por técnicas moleculares en el presente estudio, no difiere de lo que se reporta por autores que han hecho análisis similares en el país, al ser aceptado en el 88% de los casos. (21) (22) De las 144 parejas de alto riesgo, solo una rechazó el ofrecimiento de DPN por técnicas de biología molecular, en el marco del ase-soramiento genético. El resto de las que no se realizaron el DPN (12,5%), dependió de la avanzada edad gestacional y las pérdidas de embarazos. A finales de la década de los 90 en todo el país se detectaron 1268 parejas de alto riesgo, de las que solamente el 41,88% se realizó DPN (23), lo que habla a favor del incremento de la educación genética de la población cubana, favorecido por el importante salto cualitativo que se produjo en el campo de la Genética Comunitaria en Cuba, con el incremento de la cobertura de atención de los servicios de genética médica en la atención primaria, con la creación de Centros de Genética Comunitaria en todos los municipios del país y la presencia de un máster en ciencias en Asesoramiento Genético en cada una de las áreas de salud de todo el país (21).
El comportamiento respecto a la decisión de la pareja frente a un resultado positivo de DPN para AF también estuvo en correspondencia con lo que observado a nivel nacional en la década del 90, donde el 73,46% de las parejas con feto afectado optaron por la culminación de la gestación (23). El porcentaje observado en la provincia de Villa Clara en la presente investigación (71%) resultó similar al observado en un periodo de 6 años en la isla caribeña de Guadalupe (70%) donde los investigadores plantean que, para lograr una mayor eficacia del programa de prevención, se precisa de una identificación temprana de las parejas de riesgo, en combinación con la elevación del nivel de conciencia a nivel individual y poblacional (24).
Sin embargo, el porcentaje de aceptación de terminación de la gestación en Camerún, país del África subsahariana, fue menor (62,5%) y en los casos de madres solteras y desempleadas fue significativamente superior (25).
Para algunos autores los índices de reducción de nacimientos de niñas y niños con hemoglobinopa-tías en Cuba es sinónimo de intolerancia hacia las personas con enfermedades genéticas, aunque otros lo ven como resultados de decisiones individuales a partir del aumento de la concientización en relación con el riesgo genético (26).
El objetivo fundamental del programa cubano para la prevención de hemoglobinopatías SS y SC es la detección de parejas de alto riesgo, ya sea pre-concepcionalmente o en etapas tempranas del embarazo, de manera que estas puedan, al recibir asesoramiento genético, decidir su conducta reproductiva en relación al embarazo en curso, en dependencia de la severidad clínica, según el genotipo fetal (7) (23).
Otro objetivo, es asegurar la atención médica a los recién nacidos enfermos, ya sea por decisión de la pareja de continuar la gestación, o por no haber resultado posible la realización del diagnóstico prenatal. La atención precoz, la educación a los padres y demás familiares acerca del manejo de la enfermedad y la atención especializada, promueven una mejor calidad y esperanza de vida para estos pacientes (7) (23) (9) (13) (14).
La tasa de incidencia de hemoglobinopatías en los 25 años estudiados en la provincia de Villa Clara (1 por cada 10 000 habitantes) es 3.4 veces superior a la reportada en Estados Unidos de América, aunque las cifras en este país presentan grandes variaciones en relación a los diferentes grupos étnicos (1 por cada 500 nacidos vivos en afroamericanos y 1 por cada 1 000 - 1 400 nacidos vivos hispanoamericanos) (27).
Se estima que en la actualidad la incidencia de la enfermedad ha disminuido de manera considerable en Cuba. De alrededor de 100 nacimientos de niños enfermos anuales, en la actualidad nacen un promedio de 10 por año, en su mayoría descendientes de parejas que aunque conocían el diagnóstico en la etapa prenatal, decidieron continuar el embarazo (9).
A pesar de los grandes avances tecnológicos y terapéuticos encaminados hacia la prevención efectiva y adecuado manejo clínico de los casos, existe un incremento del número de recién nacidos afectados, con un elevada frecuencia de morbilidad y mortalidad, la mayoría en los países en desarrollo afectados. La OMS informa que anualmente nacen de 300 000 a 500 000 recién nacidos con hemoglobinopatías a nivel mundial, de ellos el 80% nace en países en desarrollo (28).
REFERENCIAS BIBLIOGRÁFICAS
1) SPEICHER MR,ANTONARAKIS SE, MOTULSKYAG. Human Genetics. Problemsand Aproaches. 4th ed. London: Springer; 2010.p.366-79. [ Links ]
2) ONLINE MENDELIAN INHERITANCE IN MAN. (Revisado 23 Ago 2013) URL disponible en: http://www.omim.org/search?index=entry&start=1&limit=10&search=sickle+cell+anemia&sort=score+d esc%2C+prefix sort+desc [ Links ]
3) LEHMANN H, HUNSTMAN RC. Men^s Haemoglobin. 2nd ed. Amsterdan: North-Holland. 1974. p. 567-72. [ Links ]
4) BRANDAN N, AGUIRRE MV, GIMÉNEZ CE. Hemoglobina. (Revisado 23 Ago 2013) URL disponible en:
http://www.med.unne.edu.ar/catedras/bioquimica/pdf/hemoglobina.pdf
5) CANTALEJO MA. Protocolo de Anemia de células falciformes o Drepanocitosis. DREP-2002-SEHP. (Revisado 23 Ago 2013) URL disponible en: http://www.svnp.es/Documen/protodrepanocitosis.html [ Links ]
6) VASCONCELOS A, PRIOR AR, FERRÃO A, MORAIS A. An adolescent with sickle cell anaemia expe-riencing disease-related complications: priapism and leg ulcer- A management challenge.BMJ Case Rep.2012; 2012. [ Links ]
7) MARTÍN MR, GRANDA H. Indicación temprana de electroforesis de hemoglobina a gestantes de Ciudad de La Habana. Rev Cubana Med Gen Integr 2000; 16(3):249-52. [ Links ]
7) MARTÍN MR, GRANDA H. Indicación temprana de electroforesis de hemoglobina a gestantes de Ciudad de La Habana. Rev Cubana Med Gen Integr 2000; 16(3):249-52.
8) MODELL B, DARLISON M.Global epidemiology of haemoglobin disorders and derived service indicators. Bull World Health Organ. 2008; 86(6):480-7 [ Links ]
9) SVARCH E, MARCHECO B, MACHÍN S, MENÉNDEZ A, NORDET I, ARENCIBIAA, ETAL. La drepa-nocitosis en Cuba. Estudio en niños. Rev Cubana Hematol Inmunol Hemoter. 2011; 27(1): 51-67 (1). [ Links ]
10) OFICINA NACIONAL DE ESTADÍSTICA E INFORMACIÓN DE LA REPÚBLICA DE CUBA. (Revisado 23 Ago 2013) URL disponible en: http://www.one.cu/publicaciones/08informacion/2010unamiradaacu-ba/09Villa%20Clara.pdf [ Links ]
11) FERRERO ME, PÉREZ MT, ALVAREZ R, RODRÍGUEZ L. Comportamiento clínico-epidemiológico de los defectos congénitos en la Ciudad de La Habana. Rev Cubana Pediatr 2005; 77. [ Links ]
12) Drepanocitosis y otras hemoglobinopatías. (Revisado 23 Ago 2013) URL disponible en: http://www.who.int/mediacentre/factsheets/fs308/es/ [ Links ]
13) FERNÁNDEZ JD, CABRERA M, ÁLVAREZ O, PRIETO L, MEDIACEJA O, VILLARES I. Comprehensive care for patients with sickle cell disease in Cuba. Haematologica. 2008; 93(1). (Revisado 23 Ago 2013) URL disponible en: http://www.haematologica.org/content/93/1/e20.full.pdf+html [ Links ]
14) SVARCH E, HERNÁNDEZ P, BALLESTER JM. La drepanocitosis en Cuba. Rev Cubana Hematol Inmunol Hemoter. 2004; 20(2) [ Links ]
15) MARTÍN MR, CASAS J. Conocimientos sobre Sicklemia y riesgo genético en portadoras sanas que habían recibido asesoramiento genético. Rev Habanera Ciencias Méd. 2006; 5(4):1-14. [ Links ]
16) AMERICAN ACADEMY OF PEDIATRICS. Sickle cell disease. (Revisado 23 Ago 2013) URL disponible en: http://www.aap.org/en-us/about-the-aap/Committees-Councils-Sections/section-hematology-oncology/Documents/SCDJactsheet.pdf [ Links ]
17) TORRES E, TORRES T, VALBUENA G, ARTEAGA M. Frecuencia de anemia falciforme en la población de "cuatro bocas", Parroquia Ricaurte, Municipio Mara, Estado Zulia, Venezuela. Invest Clin. 1993; 34(2): 99-105. [ Links ]
18) ROSERO MJ, BERMÚDEZ JA. Análisis de hemoglobinopatías en regiones afrocolombianas usando muestras de sangre seca de cordón umbilical. Acta Méd Colombiana. 2012; 37(3): 118- 24. [ Links ]
19) BATISTA A, ANDRADE TC. Anemia falciforme: um problema de saúde pública no Brasil. Universitas Ciencias de Saúde. 2012; 3(1): 83-99. [ Links ]
20) REES D, WILLIAMS T, GLADWIN M. Sickle-cell disease. Lancet 2010; 376: 2018-31 [ Links ]
21) GONZÁLEZ R, MAZA MA, OLIVA Y, MENÉNDEZ R. Resultados del programa de prevención de hemoglobinopatías SS y SC. Rev Ciencias Méd. 2013; 17(4): 44-53. [ Links ]
22) DOMÍNGUEZ M, VIÑALES MI, SANTANA ME, MORALES E. Pesquisaje y dilema del asesoramiento genético en parejas de riesgo de anemia a hematíes falciformes. Rev Cubana Med Gen Integr. 2005; 21 (1-2). [ Links ]
23) GRANDA H, GISPERT S, DORTICÓS A, MARTÍN M, CUADRAS Y, CALVO M, ET al. Cuban programme for prevention of sickle cell disease. Lancet. 1991; 337(8734): 152-3. [ Links ]
24) ALEXANDRE L, KECLARD L, ROMANA M, SAINT-MARTIN C, LAVOCAT-BERNARD E, MIDONET N, ET AL. Efficiency of prenatal counselling for sickle cell disease in Guadeloupe. Genet Couns. 1997; 8(1):25-32. [ Links ]
25) WONKAM A, MJAMNSHI A, MBANYA D, NGOGANG J, ZAMEYO C, ANGWAFO F. Acceptability of prenatal diagnosis by a sample of parents of sickle cell anemia patients in Cameroon (Sub-Saharan Africa). J Genet Counseling. 2011; 20: 476-85. [ Links ]
26) DINIZ D, GUEDES C. Anemia Falciforme: Um Problema Nosso. Uma abordagem bioética sobre a nova genetic. Cad. Saú de Pública. 2003; 19(6): 1761- 70. [ Links ]
27) Prevalence and Incidence of Sickle Cell Anemia. Incidence rate. (Revisado 23 Ago 2013) URL disponible en: httpy/www.rightdiagnosis.com/s/sickle^elLanemia/prevalence.html [ Links ]
28) WHO-TIF Meeting on the management of hemoglobin disorders. (Revisado 23 Ago 2013) URL disponible en: http://www.who.int/genomics/WHO-TIF_genetics_final.pdf [ Links ]